
Anormalidades Fetais
Seja bem-vindo a esse curso educacional sobre Anormalidades Fetais.
Ultrassonografia é a principal ferramenta diagnóstica na detecção pré-natal de anormalidades congênitas. Permite exame da anatomia interna e externa do feto e a detecção, não apenas de grandes defeitos, mas também de marcadores sutis de anormalidades cromossômicas e síndromes genéticas.
Apesar de algumas mulheres apresentarem alto risco para anomalias cromossômicas, por história familiar ou por exposição a teratógenos como infecção e drogas diversas, a grande maioria das anormalidades fetais ocorre no grupo de baixo risco. Consequentemente, exame ultrassonográfico deve ser oferecido rotineiramente a todas as gestantes.
Esse curso resume a prevalência, características ultrassonográficas pré-natais, manejo e prognóstico para anormalidades comuns assim como para as raras. Por favor note que, defeitos cardíacos não foram incluídos nesse curso pois são abordados em outro curso FMF especificamente sobre ecocardiograma fetal.
O curso dura várias horas, mas não precisa ser completado em uma única sessão.
Em completando o curso, você obterá o certificado de conclusão.
O curso é obrigatório como parte da certificação da FMF em Anormalidades Fetais.
Conteúdo do Curso
- Introdução
- Anormalidades cerebrais
- Anormalidades de coluna
- Anormalidades de face
- Anormalidades de pescoço
- Anormalidades de tórax
- Anormalidades de parede abdominal
- Anormalidades de trato gastrointestinal
- Anormalidades de trato urinário
- Anormalidades de trato genital
- Anormalidades de extremidades
- Anormalidades esqueléticas
- Hidropsia Fetal
- Defeitos Cromossômicos
- Tumores
- Anormalidades de placenta e cordão
- Anormalidades de líquido amniótico
- Anormalidades em gestações múltiplas
- Agradecimentos
Cérebro
Tópicos nessa seção
- Acrania
- Cisto Aracnóide
- Cisto da Bolsa de Blake
- Teratoma cerebral
- Displasia cerebelar
- Cisto de plexo coróide
- Agenesia de corpo caloso
- Malformação de Dandy-Walker
- Trombose de Seio Dural
- Encefalocele
- Hemimegaloencefalia
- Holoprosencefalia
- Hidranencefalia
- Lisencefalia
- Macrocefalia
- Megacisterna Magna
- Microcefalia
- Cisto porencefálico
- Esquizencefalia
- Displasia septo-óptica
- Esclerose tuberosa
- Aneurisma da veia de galeno
- Ventriculomegalia
Acrania
Prevalência:
- 1 em 1000 com 12 semanas
Diagnóstico ultrassonográfico:
- Ausêcia de caixa craniana e hemisférios cerebrais.
- Com 12 semanas é suspeitada pela ausência de ossificação normal do crânio e distorção cerebral (exencefalia). Com mais de 16 semanas o tecido cerebral já foi destruído (anencefalia).
Anormalidades associadas:
- Defeitos cromossômicos são raros em acrania isolada.
- Defeitos de SNC ou outros são vistos em 50% dos casos incluindo espinha bífida em 25%.
Investigação:
- Ultrassonografia detalhada.
Seguimento:
- Se a gravidez continuar, acompanhamento padrão.
Parto:
- Assistência obstétrica de rotina.
Prognóstico:
- Condição letal com morte dentro da primeira semana de vida. Recorrência:
- Um filho afetado: 5%
- Dois filhos afetados: 10% • Suplementação da dieta materna com folato (5mg/d) por 3 meses antes e 2 meses após a concepção reduz o risco de recorrência em aproximadamente 75%.
Cisto Aracnóide
Prevalência:
- 1 em 100 nascimentos. Entretanto, apenas 1% é grade suficiente para diagnóstico pré-natal.
Diagnóstico ultrassonográfico:
- Cistos uniloculares, avasculares que não se comunicam com ventrículos laterais.
- Usualmente, vistos na linha média entre os hemisférios cerebrais, mas em torno de 10% são na fossa posterior atrás do vermis.
Anormalidades associadas:
- Cistos aracnoides são, em geral, isolados. Há associações raras com anomalias cromossômicas, principalmente trissomia 18 ou 12, e agenesia de corpo caloso.
Investigação:
- Ultrassonografia detalhada, incluindo neurossonografia.
- RMN do cérebro fetal pode ser útil se USG sugerir presença de outras anormalidades cerebrais. Seguimento:Ultrassom a cada 4 semanas para monitorar tamanho do cisto e possível compressão resultando em ventriculomegalia.
Parto:
- Local: hospital com UTI neonatal e neurocirugia pediátrica.
- Momento: 38 semanas
- Via: Cesariana se circunferência cefálica > 40cm.
Prognóstico:
- Cisto pequeno isolado: neurodesenvolvimento normal.
- Cisto grande com efeito de massa: cirurgia é necessária para prevenção de sequela a longo prazo, incluindo, convulsões, cefaleia, déficit motor e retardo no desenvolvimento neurológico.
Recorrência:
- Não há aumento no risco.
Cisto da Bolsa de Blake
Prevalence:
- 1 em 1.000 nascimentos.
Diagnóstico ultrassonográfico:
- Expansão do 4o ventrículo para cisterna magna resultando em cisto unilocular, avascular na fossa posterior – sinal da fechadura no corte cerebelar transverso.
- Vermis: tamanho normal ligeiramente deslocado para cima.
- Cisterna magna: normal.
- Diagnóstico diferencial: mega cisterna magna (>10mm; vermis normal), cisto aracnoide (cisto na cisterna magna com efeito de massa sobre as estruturas adjacentes; vermis normal).
Anormalidades associadas:
- Em geral achado isolado.
- Risco de anomalias cromossômicas, principalmente trissomia 21, em até 5% dos casos, mas em geral com presença de outros marcadores sugestivos.
Investigação:
- Ultrassonografia detalhada, incluindo neurossonografia.
- RMN do cérebro fetal pode ser útil se USG sugerir presença de outras anormalidades cerebrais.
- Teste invasivo e array são recomendados nos casos não isolados.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar tamanho do cisto e possível compressão resultando em ventriculomegalia.
- Resolução espontânea com 24-26 semanas em 50% dos casos.
Parto:
- Assistência obstétrica de rotina.
Prognóstico:
- Neurodesenvolvimento: bom em 90% dos casos, levemente comprometido em 10%.
- Pequeno risco de hidrocefalia pós-natal com necessidade de shunt.
Recorrência:
- Isolado: não há aumento no risco.
- Parte de trissomia: 1%.
Teratoma cerebral
Prevalência:
- 1 em 1.000.000 de nascimentos.
- Teratomas são os tumores cerebrais mais comuns.
Diagnóstico ultrassonográfico:
- Massa sólida irregular com componentes císticos e/ou calcificados, distorcendo a anatomia cerebral.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada. Investigação:
- Ultrassonografia detalhada incluindo neurossonografia. Seguimento:
- Ultrassom a cada 4 semanas para monitorar o tamanho do tumor: teratoma apresentam crescimento rápido e podem estar associados a hidrocefalia progressiva e polidramnia.
Parto:
- Local: hospital com UTI neonatal e neurocirurgia pediátrica.
- Momento: 38 semanas.
- Via: Cesariana se circunferência cefálica > 40cm.
Prognóstico:
- Taxa de sobrevivência: <10%.
Recorrência:
- Não há aumento no risco.
Displasia cerebelar
Prevalência:
- 1 em 100.000 nascimentos.
Diagnóstico ultrassonográfico:
- Diâmetro transcerebelar <5o percentil para idade gestacional.
- *O cerebelo pequeno é causado por falha na substância branca ou do vermis (displasia vermiana) ou dos hemisférios cerebelares (displasia hemisférica).
Associated abnormalities:
- As 2 displasias vermianas mais comuns são:
- Síndrome de Joubert: mutação autossômica recessiva no cromossomo 9q34; ausência ou subdesenvolvimento do vermis cerebelar com fenda entre os hemisférios e comunicação entre o 4o ventrículo e a cisterna magna. A condição é associada a retardo no neurodesenvolvimento.
- Rombencefalosinapse: anormalidade esporádica; ausência completa de vermis com fusão dos hemisférios cerebelares, ausência de cavum do septo pelúcido, ventriculomegalia e desordens de migração. Condição associada a retardo severo do neurodesenvolvimento.
Investigação:
- Ultrassonografia detalhada incluindo neurossonografia.
- Teste invasivo para cariótipo e array.
- TORCH ( CMV pode causar cerebelo hipoplásico).
- RMN cerebral ?32 semanas para diagnóstico de anomalias não detectadas por USG como anormalidades de migração.
Seguimento:
- Rotineiro.
Parto:
- Assistência obstétrica de rotina, mas o parto deve ser realizado em hospital com UTI neonatal.
Prognóstico:
- Morte na infância.
Recorrência:
- Síndrome de Joubert: 25%.
- Rombencefalosinapse: não há aumento no risco.
Cisto de plexo coróide
Prevalência:
- 1 em 50 fetos com 20 semanas.
- Mais de 90% resolvem até 26 semanas.
Diagnóstico ultrassonográfico:
- Áreas císticas únicas ou múltiplas (>2mm de diâmetro) em um ou ambos os plexos coroides dos ventrículos laterais. Anormalidades associadas:
- Associados a risco aumento de trissomia 18 ou possivelmente trissomia 21.
Investigação:
- Ultrassom detalhado para avaliar presença de outros marcadores para trissomia 18 e 21. Na ausência de marcadores, não há necessidade de teste invasivo.
Seguimento:
- Rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Bom
Recorrência:
- Isolado: não há aumento no risco.
- Parte de trissomias: 1%.
Agenesia de corpo caloso
Prevalência:
- 1 em 300 nascimentos.
Diagnóstico ultrassonográfico:
- Ausência de cavum do septo pelúcido e aparência em lágrima da parte posterior do ventrículo lateral dilatado no corte transverso do cérebro ?18 semanas.
- Ausência completa ou parcial (geralmente da parte posterior) do corpo caloso no corte sagital médio.
- Curso anormal da artéria pericalosa.
- Comprimento normal do corpo caloso de 20 semanas é 18-20mm.
Anormalidades associadas:
- Anomalias cromossômicas ( trissomias 8,13 ou 18, deleções e duplicações) são vistas em 20% dos casos.
- Em cerca de 50% dos casos, há associação com qualquer uma de 200 síndromes genéticas, defeitos do SNC ( formação anormal de giros, cistos aracnoides de linha média, complexo de Dandy-Walker e encefalocele) ou defeitos em outros sistemas ( principalmente cardíaco, esquelético e geniturinário).
Investigação:
- Ultrassonografia detalhada incluindo neurossonografia.
- Teste invasivo para cariótipo e array.
- TORCH, avaliar infecção fetal.
- RMN cerebral ?32 semanas para diagnóstico de anomalias não detectadas por USG como heterotopias da matéria cinza, sulcação tardia e anomalias de migração.
Seguimento:
- Rotineiro.
Parto:
- Assistência obstétrica e parto de rotina
Prognóstico:
- Isolada: retardo de desenvolvimento neurológico em 30% dos casos.
- Outros defeitos: prognóstico pode ser pior dependendo do tipo de defeito. Recorrência: • Isolada: 3-5%
Malformação de Dandy-Walker
Prevalência:
- 1 em 30.000 nascimentos.
Diagnóstico ultrassonográfico:
- Dilatação cística do 4o ventrículo que preenche a fossa posterior e se estende para cisterna magna.
- Hipoplasia ou agenesia completa do vermis cerebelar.
Anormalidades associadas:
- Defeitos cromossômicos, principalmente trissomia 13 ou 18, são vistos em 30% dos casos.
- Síndromes genéticas ( mais comumente: Sd. Walker-Warburg, Meckel-Gruber, Aicardi, Neu-Laxova) e defeitos ( cérebro, coração, gastrointestinais, geniturinários) são vistos em > 50% dos casos.
- Ventriculomegalia severa é comum e desenvolvida pós-natal em > 80% dos casos.
Investigação:
- Ultrassonografia detalhada incluindo neurossonografia.
- Teste invasivo para cariótipo e array.
- RMN cerebral ?32 semanas para diagnóstico de anomalias de migração.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar possível desenvolvimento de ventriculomegalia severa.
Parto:
- Assistência obstétrica de rotina, mas o parto deve ser realizado em hospital com UTI neonatal.
- Cesariana deve ser realizada em caso de circunferência cefálica >40cm.
Prognóstico:
- Isolada: retardo no neurodesenvolvimento em >50% dos casos.
- Ventriculomegalia severa: taxa de mortalidade >50% e retardo no neurodesenvolvimento na maioria dos sobreviventes.
Recorrência:
- Isolada: 3-5%.
- Parte de Trissomias: 1%.
Trombose de seio dural
Prevalência:
- 1 em 200.000 nascimentos.
Diagnóstico ultrassonográfico:
- Massa avascular, supratentorial, hiperecogênica na fossa posterior acima do cerebelo, cercada por área sonolucente triangular (dilatação do seio venoso).
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada incluindo neurossonografia.
- RMN cerebral se necessário para confirmação do diagnóstico e para descrição do local e tamanho da lesão.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar evolução da lesão.
- Repetir RMN após 6-8 semanas a fim de excluir infarto ou hemorragia no parênquima cerebral.
Parto:
- Assistência obstétrica e parto de rotina
Prognóstico:
- Cabeça de tamanho normal com trombo diminuindo de tamanho indica prognóstico favorável. Manejo pós-natal é, em geral, não intervencionista.
Recorrência:
- Não há aumento no risco.
Encefalocele
Prevalência:
- 1 em 5.000 nascimentos.
Diagnóstico ultrassonográfico:
- Defeito no osso do crânio com herniação de cisto com ou sem parênquima cerebral.
- Usualmente, occipital (85%), mas pode ser parietal (15%) e, raramente, frontal.
Anormalidades associadas:
- Defeitos cromossômicos, principalmente trissomia 13 ou 18, são vistos em aproximadamente 10% dos casos.
- Defeitos cerebrais e não cerebrais e síndromes genéticas são presentes em > 60% dos casos. As síndromes genéticas mais comuns são: Sd. Meckel-Gruber ( autossômica recessiva; polidactilia, rins multicísticos, encefalocele occipital), Sd. Walker-Warburg (autossômica recessiva; tipo II lisencefalia, agenesia de corpo caloso, malformações cerebelares, catarata) e Sd Banda Amniótica (esporádica; anormalidades únicas ou múltiplas de extremidades, crânio-faciais e de tronco devido à presença de banda amniótica).
Investigação:
- Ultrassonografia detalhada.
- Teste invasivo para cariótipo e array.
Seguimento:
- Rotineiro.
Parto:
- Local: hospital com UTI neonatal e neurocirurgia pediátrica.
- Momento: 38 semanas.
- Via: cesariana com 38 semanas para evitar trauma ao tecido cerebral exposto.
Prognóstico:
- Depende do tamanho, conteúdo e localização da encefalocele.
- Mortalidade para encefalocele posterior é 50% e para meningocele posterior e encefalocele anterior é aproximadamente 20%.
- Deficiência neurológica nos sobrevivente é >50%.
Recorrência:
- Isolada: 3-5%.
- Parte de trissomias: 1%.
- Parte de condições autossômicas recessivas: 25%.
Hemimegaloencefalia
Prevalência:
- 1 em 100.000 nascimentos.
Diagnóstico ultrassonográfico:
- Há supercrescimento cerebral e ventriculomegalia em um dos hemisférios cerebrais resultando em desvio da linha média no corte transverso tradicional da cabeça fetal. O diagnóstico, em geral, é feito >2 semanas.
- A sempre anomalias de sulcação, incluindo agiria, paquigiria ou polimicrogiria.
Anormalidades associadas:
- Anomalias cromossômicas: não há aumento no risco.
- Síndromes genéticas são muito comuns:
- Síndrome de Proteus: esporádica; hemimegaloencefalia, crescimento desproporcional de mãos e pés.
- Síndrome de Klippel-Trenaunay: esporádica, hemimegaloencefalia, hemangiomatose em um ou mais membros, defeitos cardíacos, agenesia renal, atresia laríngea.
Investigação:
- Ultrassonografia detalhada incluindo neurossonografia.
- RMN cerebral especialmente para diagnóstico diferencial com tumores cerebrais.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar evolução da circunferência cefálica.
Parto:
- Assistência obstétrica e parto de rotina.
- Cesariana caso circunferência cefálica > 40cm.
Prognóstico:
- Reservado: retardo severo no neurodesenvolvimento, hemiplegia e convulsões intratáveis.
Recorrência:
- Não há aumento no risco.
Holoprosencefalia
Prevalência:
- 1 em 1.300 fetos até 12 semanas.
- 1 em 10.000 nascimentos.
Diagnóstico ultrassonográfico:
- Há 4 tipos:
– Alobar: fusão dos hemisférios cerebrais com ventrículo único.
– Semilobar: hemisférios cerebrais e ventrículos laterais fundidos anteriormente mas separados posteriormente.
– Lobar: hemisférios cerebrais separados tanto anterior quanto posteriormente, mas há fusão parcial dos cornos frontais dos ventrículos laterais, ausência de cavum septo pelúcido e anormalidades do corpo caloso, cavum e trato olfatório. O principal diagnóstico diferencial é displasia septo-óptica e, portanto, esforços devem ser feitos para examinar o quisma e nervo ópticos por RMN.
– Sintelecefalia: as áreas anterior e occipital cerebrais são completamente clivadas como no tipo lobar, mas diferentemente dessa, não há clivagem parietal e, portanto, as fissuras de Sylvius são verticalizadas e conectadas anormalmente através da linha média sobre o vértex cerebral.
- Holoprosencefalia lobar é detectada ? 18 semanas, mas os outros 3 tipos são detectados na USG 11-13 semanas.
Anormalidades associadas:
- Defeitos cromossômicos, pincipalmente 13 ou 18, são vistos em > 50% dos casos em 12 semanas.
- Síndromes genéticas são vistas em 20% dos casos.
- Holoprosencefalia alobar e lobar são associadas à microcefalia e defeitos de linha média faciais em 80% dos casos. Defeitos extracerebrais são particularmente comuns em fetos com trissomia 13 e 18 e naqueles com síndromes genéticas.
Investigação:
- Ultrassonografia detalhada incluindo neurossonografia.
- Teste invasivo para cariótipo e array.
- RMN cerebral pode ser útil para confirmação diagnóstica em casos suspeitos de holoprosencefalia lobar.
Seguimento:
- Se a gravidez continuar, acompanhamento padrão.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Alobar e semilobar: usualmente letais dentro do 1o ano de vida. • Lobar: expectativa de vida pode ser normal mas, usualmente, grave retardo no desenvolvimento e alterações visuais.
Recorrência:
- Isolada: 6%.
- Parte de trissomias: 1%.
- Parte de síndromes genéticas: 25-50%.
Hidranencefalia
Prevalência:
- 1 em 50.000 nascimentos.
Diagnóstico ultrassonográfico:
- A cavidade craniana é repleta de líquido e não há córtex restante. A foice cerebral e fossa posterior são normais. Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
- As causas são oclusão vascular da artéria carótida interna, infecção fetal ou ventriculomegalia prolongada.
Investigação:
- Ultrassonografia detalhada, incluindo neurossonografia.
- TORCH para rastreio de infecção fetal.
Seguimento:
- Se gravidez continuar, acompanhamento padrão.
Parto:
- Assistência obstétrica e parto de rotina.
- Se circunferência cefálica >40cm, cefalocentese pode ser necessária antes de parto vaginal.
Prognóstico:
- Morte nos primeiros anos da infância.
Recorrência:
- Não há risco aumentado.
Lisencefalia
Prevalência:
- 1 em 100.000 nascimentos.
Diagnóstico ultrassonográfico:
- Todo ou parte do cérebro aparenta ser liso, com ausência de desenvolvimento de giros e sulcos.
- Diagnóstico pré-natal é muito difícil, mas suspeita pode ser levantada >24 semanas. As fissuras parieto-occipital e de Sylvius aparentam ser planas e o espaço subaracnóide é, em geral, aumentado.
- Em todos os casos de anomalias cerebrais aparentemente isoladas, como ventriculomegalia e agenesia de corpo caloso, USG detalhada por volta de 26 semanas deve ser realizada para excluir lisencefalia subjacente.
- Há 2 tipos de lisencefalia:
- Tipo I: agiria com falta de migração neuronal. O córtex é liso e grosso/espessado.
- Tipo II: migração extensiva e anárquica com falta de camadas. O córtex é descrito como em cobblestone.
Anormalidades associadas:
- Anomalias cromossômicas: não há risco aumentado.
- A condição pode ocorrer isolada, mas está comumente associada a síndromes genéticas
- Miller-Dieker: esporádica; lisencefalia tipo I, microcefalia, defeitos cardíacos e polidactilia.
- Walker-Warburg: autossômica recessiva; lisencefalia tipo II, agenesia de corpo caloso, malformações cerebelares, catarata.
Investigação:
- Ultrassonografia detalhada, incluindo neurossonografia.
- RMN cerebral ? 32 semanas para diagnóstico de desordens de migração neuronal.
- Cariótipo fetal: para diagnóstico da síndrome de Miller-Dieker ( deleção 17p13.3)
- Cariótipo dos pais: para diagnóstico de possível translocação balanceada envolvendo 17p.
Seguimento:
- Avaliação a cada 4 semanas.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Expectativa de vida é de aproximadamente 10 anos com grave retardo no desenvolvimento neurológico.
Recorrência:
- Translocação nos pais: 75%
- Isolada: não há aumento no risco.
Macrocefalia
Prevalência:
- 1 em 100 nascimentos.
- Mais comum em homens que mulheres: 4:1.
Diagnóstico ultrassonográfico:
- Circunferência cefálica > 2 desvios-padrão (DP).
Anormalidades associadas:
- Tipos de macrocefalia:
- Familiar (maioria): benigna, neurodesenvolvimento normal.
- Secundária: devido a condições subjacentes (ex.: hidrocefalia, tumores cerebrais)
- Megaloencefalia: desordem de proliferação neuronal e da glia que causa supercrescimento de células resultando em retardo do neurodesenvolvimento grave. Mais de 100 síndromes com supercrescimento pré e pós-natal foram descritas. A mais comum é a Sd. Sotos (autossômica dominante, mas 95% dos casos são devido a mutação nova – de novo; macrocefalia, bossa frontal e hipertelorismo).
Investigação:
- Ultrassonografia detalhada, incluindo neurossonografia.
- RMN cerebral.
- Teste invasivo para cariótipo e array. Megaloencefalia é associada a grandes deleções em vários cromossomos assim como microdeleções.
Seguimento:
- Acompanhamento padrão.
Parto:
- Assistência obstétrica e parto de rotina. • Via: cesariana caso circunferência cefálica >40cm.
Prognóstico:
- Familiar: neurodesenvolvimento normal.
- Secundária: depende da patologia de base.
- Unilateral ou Bilateral: retardo no desenvolvimento e convulsões intratáveis.
Recorrência:
- Familiar (padrão de herança autossômico dominante): 50%.
- Secundária: não há aumento no risco.
- A maioria das síndromes tem padrão de herança autossômico dominante, mas mais de 95% são devido a mutações novas.
Megacisterna magna
Prevalência:
- 1 em 5.000 nascimentos.
Diagnóstico ultrassonográfico:
- A cisterna magna é > 10mm no corte cerebelar transverso.
- Vermis: normal.
- Diagnóstico diferencial: Cisto da Bolsa de Blake (expansão do 4o ventrículo em direção à cisterna resultando em cisto avascular, unilocular na fossa posterior; vermis normal deslocado superiormente), cisto aracnoide (cisto na cisterna magna com efeito de massa nas estruturas adjacentes; vermis normal).
Anormalidades associadas:
- Em geral, achado isolado, mas em até 10% dos casos há ventriculomegalia.
Investigação:
- Ultrassonografia detalhada, incluindo neurossonografia.
- RMN cerebral pode ser útil caso outras anomalias sejam suspeitadas.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar tamanho da cisterna e possível desenvolvimento de ventriculomegalia.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Neurodesenvolvimento normal.
Recorrência:
- Não há aumento no risco.
Microcefalia
Prevalência:
- 1 em 1.000 nascimentos.
- 80% das crianças afetadas tem circunferência cefálica normal ao nascimento e 90% teve a medida da CC normal no segundo trimestre.
Diagnóstico ultrassonográfico:
- Diagnóstico ultrassonográfico é feito, em geral, no final do segundo e no terceiro trimestres.
- A razão da circunferência cefálica/circunferência abdominal está abaixo do percentil 3 ( 2 DP abaixo do normal para idade gestacional).
- A teste é inclinada devido à desproporção dos lobos frontais em relação a face.
- Em vários casos diagnosticados no segundo trimestre, há holoprosencefalia ou encefalocele associadas e nos diagnosticados apenas no terceiro trimestre, há anormalidades de sulcação e migração neuronal.
Anormalidades associadas:
- Anomalias cromossômicas são raras e as trissomias mais comuns são 13, 18 e 21.
- Síndromes genéticas são muito comuns, a maioria sendo causada por defeitos em único gene com padrão de herança ou autossômico recessivo ou ligado ao X. As mais comuns são: Meckel-Gruber, Walker-Walburg, Miller-Diecker, Smith-Lemil-Optiz e Seckel.
Investigação:
- Ultrassonografia detalhada, incluindo neurossonografia.
- Teste invasivo para cariótipo e array.
- TORCH
- RMN cerebral ? 32 semanas para diagnóstico de desordens de migração neuronal como lisencefalia e polimicrogiria.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar evolução da circunferência cefálica.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Isolada: o risco de retardo no neurodesenvolvimento aumenta inversamente proporcional a CC, de 10% com a circunferência 2/3 DP abaixo do normal para idade gestacional, para 100% se > 4DP.
- Sindrômico: o prognóstico depende da condição subjacente.
Recorrência:
- Sem associação com defeitos estruturais: 5-10%
- Forma Familiar Isolada: 25%.
Cisto porencefálico
Prevalência:
- 1 em 10.000 nascimentos.
Diagnóstico ultrassonográfico:
- Uma ou mais lesões císticas no cérebro que se comunicam com os ventrículos, espaço subaracnóide ou ambos. Os cistos são ou nas fissuras, ou na linha média.
- Há 2 tipos de porencefalia:
- Tipo I: unilateral, devido à hemorragia ou isquemia.
- Tipo II: bilateral: devido à defeito de migração neuronal.
Anormalidades associadas:
- Incidência de defeitos cromossômicos não é aumentada.
- Defeitos estruturais, principalmente, ventriculomegalia.
Investigação:
- Ultrassonografia detalhada, incluindo neurossonografia.
- TORCH
- RMN cerebral ? 32 semanas para diagnóstico de heterotopias de matéria cinza, sulcação tardia e anomalias de migração.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar evolução da lesão e de ventriculomegalia.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Depende do tamanho e localização da lesão, mas o neurodesenvolvimento é, em geral, normal.
Recorrência:
- Não há aumento do risco.
Esquizencefalia
Prevalência:
- 1 em 100.000 nascimentos.
Diagnóstico ultrassonográfico:
- Fenda uni ou bilateral entre o sistema ventricular e o espaço subaracnóide.
- Em 70% dos casos a lesão é no lobo parietal.
- Em 50-90% dos casos há outras anormalidades associadas, incluindo agenesia de cavum septo pelúcido, displasia septo-óptica e ventriculomegalia grave.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada, incluindo neurossonografia.
- RMN cerebral é necessária para distinguir esquizencefalia, que é desordem de migração, de porencefalia, que ocorre devido a insulto vascular. Também é importante para detecção de polimicrogiria ( excessivas circunvoluções cerebrais), achados constante na esquizencefalia.
Seguimento:
- Rotineiro
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Reservado: grave retardo no desenvolvimento e epilepsia intratável. Além disso há cegueira, caso a condição seja associada a displasia septo-óptica.
Recorrência:
- Não há risco aumentado.
Displasia septo-óptica
Prevalência:
- 1 em 10.000 nascimentos.
Diagnóstico ultrassonográfico:
- Ausência de cavum septo pelúcido com cornos anteriores comunicantes.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
- Na displasia septo-óptica, além da ausência do cavum septo pelúcido, há hipoplasia dos nervos ópticos e/ou anormalidades hipotálamo/pituitárias.
Investigação:
- Ultrassonografia detalhada, incluindo neurossonografia.
- RMN cerebral para avaliar nervos e quiasma óptico, hipotálamo e glândula pituitária e para demonstrar possíveis anomalias corticais associadas (polimicrogiria e esquizencefalia).
Seguimento:
- Rotineiro
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Ausência isolada de cavum septo pelúcido: usualmente assintomática
- Displasia septo-óptica: distúrbios visuais desde cegueira até visão quase normal. Insuficiências hormonais podem ser tratadas com terapia de reposição hormonal.
Recorrência:
- Não há aumento no risco.
Esclerose tuberosa
Prevalência:
- 1 em 6.000 nascimentos.
Diagnóstico ultrassonográfico:
- Múltiplos nódulos ecogênicos no coração (rabdomioma, usualmente > 20 semanas) e cérebro (tubérculos corticais e nódulos subependimais, usualmente > 30 semanas).
- Diagnóstico diferencial: fibroma cardíaco, que é único, grande e geralmente associado a derrame pericárdico.
- Esclerose tuberosa é vista em 50% dos casos de rabdomioma ( nos outros 50% dos casos o tumor cardíaco é isolado). Quando há múltiplos rabdomiomas, o risco de esclerose tuberosa é > 90%.
Anormalidades associadas:
- Mutações nos genes TSC1 ou TSC2 são vistas em 90% dos casos.
- Os tumores em geral afetam cérebro, coração, pele, rins, olhos e pulmões. Entretanto, o diagnóstico pré-natal é limitado à detecção de lesões no coração e cérebro.
Investigação:
- Ultrassonografia detalhada, incluindo neurossonografia.
- Pesquisa de mutações nos genes TSC1 e TSC2.
- RMN cerebral pode ser útil caso outras anormalidades sejam suspeitadas.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar evolução de tumores cardíacos e desenvolvimento de arritmias e hidropsia.
Parto:
- Local: hospital com UTI neonatal.
- Momento: 38 semanas. Antes disso caso arritmias ou hidropsia.
- Via: cesariana em caso de hidropsia.
Prognóstico:
- Arritmias, hidropsia e óbito fetal ocorrem em 20% dos casos.
- Os tumores cardíacos geralmente regridem nos primeiros anos de vida, enquanto os cerebrais geralmente aumento em número e tamanho.
- Prognóstico depende do número, tamanho e localização dos tumores.
- Um amplo espectro, desde expectativa de vida normal com sintomas leves até grave retardo no neurodesenvolvimento, epilepsia, autismo, falência renal e pulmonar.
Recorrência:
- Autossômica dominante: 50% se um dos pais afetados.
- Mutação nova – de novo – (65% dos caso): não há aumento no risco.
Aneurisma da veia de galeno
Prevalência:
- 1 em 25.000 nascimentos
Diagnóstico ultrassonográfico:
- Cisto anecóico supra-tentorial em linha média, alongado com fluxo arteriovenoso ativo demonstrado pelo color Doppler.
- O defeito se desenvolve no início do primeiro trimestre, mas o aneurisma só se torna aparente à ultrassonografia no terceiro trimestre.
- Em 90% dos casos há insuficiência cardíaca de alto débito e hidropsia secundária.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
Investigação:
- Neurossonografia para excluir lesão cerebrais secundárias ( isquemia e leucomalácia).
- Ecocardiograma para detecção de sinais precoces de insuficiência cardíaca.
Seguimento:
- Ultrassom semanal com o objetivo principal de determinar momento de interrupção que depende do risco de desenvolvimento de insuficiência cardíaca.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: Cesariana.
Prognóstico:
- Um terço tem bom desfecho após intervenção neonatal (embolização). Um terço morre durante o procedimento e um terço sobrevive, porém com retardo no neurodesenvolvimento.
- Se houver hidropsia fetal no momento do diagnóstico o prognóstico é ruim.
Recorrência:
- Não há aumento no risco.
Ventriculomegalia
Prevalência:
- 1 em 100 fetos com 20 semanas • 1 em 1.000 nascimentos.
Diagnóstico ultrassonográfico:
- Dilatação uni ou bilateral dos ventrículos laterais cerebrais no corte transverso cerebral.
- Subdividido de acordo com o diâmetro do ventrículo lateral em leve (10-12mm), moderada (13-15mm) e severa (>15mm).
Anormalidades associadas:
- Defeitos cromossômicos, principalmente trissomias 21, 18 ou 13, são vistas em 10% dos casos. Em ventriculomegalia isolada, há um risco 4 vezes maior de trissomia 21. O risco é inversamente proporcional a severidade da ventriculomegalia.
- Defeitos cerebrais e extra-cerebrais e síndromes genéticas são vistos em 50% dos casos.
Investigação:
- Ultrassonografia detalhada, incluindo neurossonografia.
- Teste invasivo para cariótipo e array.
- TORCH • Pesquisa de anticorpos antiplaquetários no sangue materno em caso de evidência de hemorragia cerebral.
- RMN cerebral ? 32 semanas para diagnóstico de desordens de migração neuronal como lisencefalia.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar evolução da ventriculomegalia.
Parto:
- Assistência obstétrica e parto de rotina.
- Cesariana caso circunferência cefálica > 40cm.
Prognóstico:
- Isolada leve/moderada: retardo no neurodesenvolvimento em 10% dos casos, podendo não ser maior do que a população geral.
- Isolada grave: taxa de sobrevida em 10 anos de 60%, retardo mental grave em 50%.
Recorrência:
- Isolada <1%. Aumenta para 5% se há história de irmão afetado.
- Associada à infecção: não há risco aumentado.
- Associada a trissomias: 1%
- Hidrocefalia ligada ao X: 50% em homens.
- Associada à trombocitopenia autoimune sem tratamento: 100%.
Coluna
Tópicos nessa seção
- Hemivértebra
- Espinha bífida aberta
- Teratoma sacrococcígeo
Hemivértebra
Prevalência:
- 1 em 200 nascimentos.
- Mais comum em mulheres do que homens: 3:1.
Diagnóstico ultrassonográfico:
- Após 12 semanas: a coluna é distorcida nos cortes sagital e coronal, resultando em escoliose.
- A hemivértebra aparece como estrutura óssea, triangular, menor que uma vértebra normal, atuando como calço sobre vértebras normais adjacentes.
Anormalidades associadas:
- A incidência de anomalias cromossômicas não é aumentada.
- Síndromes associadas são comuns: Jarcho-Levin (autossômica recessiva; vétebras fundidas, escoliose, alinhamento de costelas anormal), Klippel-Feil ( autossômica recessiva ou dominante; fusão de vertebras cervicais), Associação VACTERL (esporádica; defeitos vertebrais e de septo ventricular, atresia anal, fístula traqueoesofágica, anomalias renais, displasia radial e artéria umbilical única), Complexo OEIS (esporádico; onfalocele, extrofia cloacal, anus imperfurado e defeitos de coluna). • Anomalias estruturais, principalmente músculo-esqueléticas, geniturinárias e cardíacos são vistos em > 70% dos casos.
Investigação:
- Ultrassonografia detalhada.
- Ultrassom 2D e 3D da coluna para definir extensão da hemivértebra e excluir lesão de cordão medular.
Seguimento:
- Ultrassom a cada 4-6 semanas para monitorar evolução da escoliose.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Isolada: bom. Em 75% dos casos não há progressão da escoliose ou essa ocorre lentamente, porém 25% apresenta rápida progressão aos 2-3 anos de idade.
- Associada a outros defeitos estruturais: reservado com alto risco de morte neonatal.
Recorrência:
- Vértebra única, isolada: não há risco aumentado.
- Múltiplas vértebras, isoladas: 5-10%
- Parte de condição autossômica recessiva: 25%.
Espinha bífida aberta
Prevalência:
- 1 em 1.000 fetos com 12 semanas.
Diagnóstico ultrassonográfico:
- Diagnóstico de espinha bífida requer exame sistemático de cada arco neural, desde a região cervical até sacral, tanto no corte transversal como longitudinal. No corte transverso, o arco neural normal aparece como um círculo fechado com pele íntegra cobrindo, enquanto que nos casos de espinha bífida, o arco tem forma de U e há meningocele abaulada adjacente (cisto de parede fina) ou mielomenigocele. A extensão da lesão e de qualquer cifoescoliose associada é melhor avaliada no corte sagital.
- Localização da espinha bífida aberta: lombossacral (65%), sacral (24%), toracolombar (10%), cervical (1%).
- Espinha bífida aberta é associada à Malformação de Arnold-Chiari II com deslocamento caudal do tronco cerebral (brain stem – BS) e obliteração da cisterna magna.
- Com 11-13 semanas, no corte sagital médio da cabeça, a parte inferior do cérebro entre o osso esfenoide anteriormente e o osso occipital (occipital bone – OB) posteriormente, pode ser dividida em tronco cerebral anteriormente e uma combinação de 4o ventrículo e cisterna magna posteriormente. Na maioria dos casos, o diâmetro do tronco está aumentado, a distância entre o tronco e o osso occipital (“BSOB”) está diminuída e a razão BS/BSOB é >1.0 .
- No segundo trimestre, mais de 95% dos fetos apresentam acavalgamento dos ossos frontais ( sinal do limão) e obliteração da cisterna magna com “ausência” de cerebelo ou curvatura anormal dos hemisférios cerebelares ( sinal da banana). Grau variável de dilatação ventricular está presente em todos os casos de espinha bífida ao nascimento, mas em apenas 70% dos casos no segundo trimestre.
Anormalidades associadas:
- Anomalias cromossômicas (principalmente trissomia 18), genes mutantes únicos e diabetes materno ou ingestão de drogas antiepiléticas estão ligados a aproximadamente 10% dos casos.
- Risco de síndromes não cromossômicas é baixo.
Investigação:
- Ultrassonografia detalhada.
- Cariótipo fetal na presença de anomalias associadas.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar evolução de ventriculomegalia e possível escoliose, além do desenvolvimento de pé torto.
Terapia Fetal:
- Fechamento intrauterino de espinha bífida ( por cirurgia aberta com histerotomia ou por fetoscopia) reduz a necessidade de shunt pós-natal e pode melhorar desenvolvimento motor e função urológica.
Parto:
- Assistência obstétrica e parto de rotina. Não há evidência de que cesariana reduza o risco de deficiência.
Prognóstico:
- A taxa de mortalidade em 5 anos é aproximadamente 35% com 20% morrendo nos primeiros 12 meses de vida. Aproximadamente 25% dos fetos com espinha bífida são natimortos.
- As crianças sobreviventes, em geral, tem paralisia de membros inferiores e incontinência dupla; apesar de hidrocefalia associada que requer cirurgia, a inteligência, em geral, é normal.
Recorrência:
- Pai/Mãe ou 1 filho afetado: 5%
- Dois filhos afetados: 10%
- Suplementação da dieta materna com folato (5mg/d) por 3 meses antes e 2 meses após a concepção reduz o risco de recorrência em aproximadamente 75%.
Teratoma sacrococcígeo
Prevalência:
- 1 em 20.000 nascimentos.
- É o teratoma fetal mais frequente.
Diagnóstico ultrassonográfico:
- Geralmente sólido e cístico ( múltiplos cistos irregulares em forma e tamanho).
- O tumor pode ser completamente externo, parcialmente interno e parcialmente externo ou principalmente interno.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
- Hidronefrose devido a compressão ureteral em caso de tumores internos.
- Tumores grandes podem resultar em anemia fetal e trombocitopenia ( devido a sequestro de hemácias e plaquetas pelo tumor), insuficiência fetal, hidropsia e placentomegalia ( devido à circulação hiperdinâmica como resultado de shunt arteriovenoso), polidramnia ( devido a transudação direta para cavidade amniótica e poliúria fetal secundária à circulação hiperdinâmica) e síndrome espelho materna ( sobrecarga hídrica e pré-eclâmpsia).
Investigação:
- Ultrassonografia detalhada, incluindo ecocardiograma para avaliação de função cardíaca e medida do pico de velocidade sistólica da artéria cerebral média (PVS ACM) para diagnóstico de anemia fetal.
Seguimento:
- Ultrassom a cada 2-3 semanas para monitorar crescimento do tumor, função cardíaca, PVS ACM e volume de líquido amniótico.
- Coagulação a laser de vasos tumorais guiada por ultrassom, transfusão intrauterina e amniodrenagem podem se tornar necessários.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas. Mais precocemente se houver evidência de restrição de crescimento, hipóxia fetal ou hidropsia.
- Via: cesariana se tumor muito grande e houver evidência de insuficiência cardíaca.
Prognóstico:
- Mortalidade perinatal: aproximadamente 50%, principalmente devido a parto prematuro ( consequência da polidramnia) de feto hidrópico que requer cirurgia neonatal de grande porte.
- Cirurgias complicadas, especialmente com tumores que se estendem para pelve e abdômen, podem resultar em lesão nervosa e incontinência.
- O tumor é invariavelmente benigno no período neonatal mas o atraso na cirurgia ou remoção incompleta pode resultar em transformação maligna ( aproximadamente 10% antes de 2 meses de idade e 80% até 4 meses).
Recorrência:
- Não há risco aumentado.
Face
Tópicos nessa seção
- Anoftalmia
- Catarata
- Dacriocistocele
- Epignathus
- Fenda facial
- Hipertelorismo
- Hipotelorismo
- Micrognatia
- Anomalias nasais
Anoftalmia
Prevalência:
- 1 em 20.000 nascimentos.
Diagnóstico ultrassonográfico:
- Na microftalmia, há diminuição do tamanho do globo ocular e na anoftalmia, há ausência do globo, nervo e quiasma ópticos. Ambas podem ser uni ou bilaterais.
Anormalidades associadas:
- Defeitos cromossômicos, principalmente trissomia 13, são vistos em > 50% dos casos.
- Síndromes genéticas estão presentes em >50% dos casos. As mais comuns são: Sd. Goldenhar (esporádica; anoftalmia, defeitos de orelha, fenda e macrossomia faciais), Sd. Fraser (autossômica recessiva; microftalmia, fenda facial, atresia traqueal, agenesia renal bilateral, defeitos cardíacos, sindactilia ou polidactilia), Sd. Fryns ( autossômica recessiva; anoftalmia, fenda facial, micrognatia, ventriculomegalia, hérnia diafragmática).
Investigação:
- Ultrassonografia detalhada, incluindo neurossonografia.
- Teste invasivo para cariótipo e array.
- RMN cerebral para diagnóstico de anormalidades como ausência de nervo óptico.
Seguimento:
- Nos casos isolados acompanhamento rotineiro. Caso haja síndrome subjacente, assistência antenatal deve ser ajustada conforme o risco de cada condição.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Isolada: bom.
- Sindrômica: reservado.
Recorrência:
- Isolada: não há riso aumentado
- Parte da trissomia 13: 1%.
- Parte de condição autossômica recessiva: 25%.
Catarata
Prevalência:
- 1 em 10.000 nascimentos.
Diagnóstico ultrassonográfico:
- Opacidade uni ou bilateral dos cristalinos.
- Acometimento bilateral, em geral, é sindrômico, enquanto unilateral, usualmente é relacionado à infecção fetal.
Anormalidades associadas:
- A incidência de defeitos cromossômicos não é aumentada.
- Síndromes genéticas são vistas em 10% dos casos. As mais comuns incluem: Walker-Warburg ( autossômica recessiva; lisencefalia tipo II, agenesia de corpo caloso, malformações cerebelares, catarata) e Condrodisplasia punctata ( recessiva ligada ao X; catarata, encurtamento rizomélico simétrico e calcificações de epífise).
- Infecção congênita ( especialmente rubéola, toxoplasmose e CMV) vista em 30% dos casos.
Investigação:
- Ultrassonografia detalhada, incluindo neurossonografia.
- Teste invasivo para cariótipo e array.
- TORCH
Seguimento:
- Rotineiro nos casos isolados.
- Em casos suspeitos de infecção, ultrassom a cada 2 semanas.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Isolada: em geral, bom. Intervenção oftalmológica após nascimento tem bons resultados, não afetando a qualidade de vida.
- Sindrômica: em geral, ruim.
Recorrência:
- Isolada: não há risco aumentado.
- Parte de síndromes: 25%.
Dacriocistocele
Prevalência:
- 1 em 4.000 nascimentos.
Diagnóstico ultrassonográfico:
- Cisto (75% unilateral e 25% bilateral) entre parte inferior da órbita e o nariz.
- Aproximadamente 90% das dacriocistoceles ocorrem devido à canalização tardia do ducto lacrimal após 32 semanas.
- Diagnóstico diferencial inclui encefalocele anterior ( geralmente associadas a anormalidades intracranianas como hidrocefalia), hemangiomas (usualmente sólidos ou multiseptados) e cistos dermóides ( em geral, localizados súpero-lateralmente).
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada.
Seguimento:
- Rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Resolução espontânea ou no terceiro trimestre ou nos primeiros 6 meses de vida.
- Ocasionalmente, sondagem do ducto nasolacrimal pode ser necessária para desobstrução.
Recorrência:
- Não há risco aumentado.
Epignathus
Prevalência:
- 1 em 200.000 nascimentos.
Diagnóstico ultrassonográfico:
- Tumor sólido que emerge do osso esfenoide, palatos duro e mole, faringe, língua e mandíbula.
- O tumor cresce em direção a cavidade oral ou nasal ou intracraniana.
- Diagnóstico diferencial inclui teratoma de pescoço, encefalocele e tumores de estruturas faciais.
- Polidramnia (devido a compressão faríngea) é usualmente presente.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada, incluindo neurossonografia.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar crescimento do tumor e avaliar volume de líquido amniótico. Em caso de polidramnia, monitoramento de comprimento de colo semanal deve ser realizado para determinar necessidade de amniodrenagem.
- RMN com 32 semanas para avaliar relação com estruturas adjacentes.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via cesariana com procedimento EXIT.
Prognóstico:
- Depende do tamanho da lesão e envolvimento de estruturas vitais.
- Polidramnia é associada a prognóstico ruim. A principal causa de morte neonatal é asfixia por obstrução de via aérea.
- Ressecção cirúrgica com pós-operatório normal é possível.
Recorrência:
- Não há risco aumentado.
Fenda facial
Prevalência:
- 1 em 700 nascimentos.
- Mais comum em homens que mulheres e em brancos do que negros. • Em 50% dos casos, tanto lábios quanto palato são afetados, em 25% apenas lábios e em 25% apenas palato.
- Unilateral em 75% dos casos (mais comum à esquerda) e bilateral em 25%.
Diagnóstico ultrassonográfico:
- A típica fenda labial aparece como defeito linear que se estende de um lado do lábio até a narina. Fenda palatina associada à labial pode se estender através do rebordo alveolar e palato duro, atingindo a base da cavidade nasal ou até o assoalho da órbita.
- Cortes transverso e coronal são necessários para diagnóstico. Doppler colorido pode ser útil para demonstrar fluxo através do palato em caso de fenda palatina.
- Diagnóstico de fenda palatina isolada é difícil.
- Diagnóstico de fenda labial e palatina entre 11-13 semanas pode ser realizado através de exame direcionado do triângulo retronasal em corte coronal e observação de falha no maxilar no corte sagital médio da face rotineiramente usado pra rastreio de anormalidades cromossômicas.
Anormalidades associadas:
- Anomalias cromossômicas, principalmente trissomia 13 e 18, são vistas em 1-2% dos casos. Fenda labial unilateral não está associada a anormalidades cromossômicas.
- Associada a quaisquer de >400 síndromes em 30% dos casos. As mais comuns são: Sd. Goldenhar (esporádica; anoftalmia, defeitos de orelha, fenda e macrossomia faciais), Sd. Treacher-Collins (autossômica recessiva ou dominante com 60% de mutações novas – de novo; hipoplasia da maxila e osso zigomático, micrognatia fenda palatina, orelhas ausentes ou malformadas), Anomalia Pierre-Robin (micrognatia ou retrognatia, fenda palatina ou glossoptose. Em metade dos casos é achado esporádico isolado e na outra metade é associado a outras anomalias ou síndromes genéticas ou não genéticas reconhecidas).
Investigação:
- Ultrassonografia detalhada.
- Teste invasivo para cariótipo e array.
Seguimento:
- Rotineiro.
- Consulta pré-natal com equipe multidisciplinar.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Depende primariamente da presença de outras anomalias e seu tipo.
- Isolada: bom com sobrevida normal.
- Reparo cirúrgico ocorre aos 3-6 meses de idade.
- Complicações a longo prazo em crianças com fenda labial e palatina incluem anormalidades dentárias, problemas auditivos e olfatórios, hipoplasia de face problemas psicológicos. Cerca de 25% tem problemas de fala necessitando de uma segunda cirurgia no palato e fonoterapia. Anomalias dentárias incluem dentes ausentes, extra ou mal posicionados e necessitam de aparelhos. A maioria das crianças tem problemas auditivos e podem necessitar de meringotomia com colocação bilateral de tubos de drenagem por timpanotomia para melhorar audição. Avaliação psicológica de rotina é recomendada para monitorar desenvolvimento cognitivo, comportamento e auto-percepção.
Recorrência:
- Isolada: 5% caso um dos pais ou filho afetado e 10% caso 2 filhos afetados.
- Sindrômica: todas as formas de herança foram descritas, incluindo autossômica dominante e recessiva, ligada ao X dominante e recessiva.
Hipertelorismo
Prevalência:
- 1 em 20.000 nascimentos.
Diagnóstico ultrassonográfico:
- Diâmetro interorbitário acima do percentil 95.
Anormalidades associadas:
- Defeitos cromossômicos, principalmente trissomia 13, são muito raros.
- Síndromes genéticas são vistas em 50% dos casos. As mais comuns são displasia frontonasal (esporádica; hipertelorismo, fenda facial mediana, anomalias de nariz e crânio biffidum ocultum), craniossinostose ( incluindo Apert, Carpenter, Crouzon) e Sd. Neu-Laxova ( autossômica recessiva; hipertelorismo, microcefalia, agenesia de corpo caloso, contraturas de membros superiores e inferiores e restrição de crescimento fetal).
- Defeitos associados: encefalocele frontal e agenesia de corpo caloso.
Investigação:
- Ultrassonografia detalhada.
- Teste invasivo para cariótipo e array.
Seguimento:
- Isolado: rotineiro.
- Sindrômico: cuidados pré-natais devem ser ajustados de acordo com os riscos de cada condição.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Isolado: em geral bom, mas em casos graves as implicações estéticas são importantes. A visão binocular esteroscópica pode ser comprometida.
- Sindrômico: em geral ruim com alto risco de retardo no neurodesenvolvimento.
Recorrência:
- Isolado: não há risco aumentado.
Hipotelorismo
Prevalência
- 1 em 20.000 nascimentos.
Diagnóstico ultrassonográfico:
- Diâmetro interorbitário abaixo do percentil 5. Hipotelorismo é parte dos defeitos de migração de linha média assim como a holoprosencefalia (que está quase sempre presente). O grau de hipotelorismo pode ser extremo, chegando a ciclopia.
Anormalidades associadas:
- Anomalias cromossômicas, principalmente trissomia 13, são vistas em 50% dos casos.
- Síndromes genéticas são muito frequentes e as mais comuns são Sd. Meckel-Gruber ( autossômica recessiva; condição letal caracterizada por encefalocele occipital, rins multicísticos e polidactilia pós-axial).
Investigação:
- Ultrassonografia detalhada, incluindo neurossonografia.
- Teste invasivo para cariótipo e array.
Seguimento:
- Rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Parte da Trissomia 13: letal.
- Cariótipo normal: alto risco de retardo no neurodesenvolvimento dependendo do grau de holoprosencefalia.
Recorrência:
- Isolado: não há risco aumentado.
- Parte da Trissomia 13: 1%.
- Parte de condição autossômica recessiva: 25%.
Micrognatia
Prevalência:
- 1 em 1.500 nascimentos.
Diagnóstico ultrassonográfico:
- Achados subjetivo de lábio superior proeminente e queixo recuado no corte sagital médio da face. Esse achado pode ser devido à micrognatia ( mandíbula curta) ou retrognatia ( deslocamento posterior da mandíbula).
- Micrognatia severa é associada a polidramnia ( >25 semanas) devido à glossoptose ( língua de tamanho normal, obstruindo cavidade oral pequena).
Anormalidades associadas:
- Anomalias cromossômicas, principalmente trissomia 18 e triploidia, são vistas em aproximadamente 30% dos casos.
- Síndromes genéticas são associadas me > 50% dos casos, incluindo:
- Anomalia Pierre-Robin: micrognatia ou retrognatia, fenda palatina ou glossoptose. Em metade dos casos é achado esporádico isolado e na outra metade é associado a outras anomalias ou síndromes genéticas ou não genéticas reconhecidas.
- Sd. Treacher-Collins: autossômica recessiva ou dominante com 60% de mutações novas – de novo; hipoplasia da maxila e osso zigomático, micrognatia fenda palatina, orelhas ausentes ou malformadas.
- Otocefalia: esporádica; micrognatia severa ou agnatia e defeitos de linha média, incluindo holoprosencefalia, encefalocele anterior, ciclopia, aglosia e localização de orelhas no meio da face.
Investigação:
- Ultrassonografia detalhada.
- Teste invasivo para cariótipo e array.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar crescimento é líquido amniótico.
Parto:
- Local: hospital com UTI neonatal.
- Momento: 38 semanas.
- Via: indução de parto vaginal. Um pediatra deve estar presente na sala de parto, preparado para entubar o neonato.
Prognóstico:
- Mortalidade neonatal: >80% por conta de anomalias associadas.
- Na anomalia Pierre-Robin a sobrevida é boa.
Recorrência:
- Isolada: não há risco aumentado.
- Parte de trissomia: 1%.
- Parte de síndromes genéticas: 25-50%.
Anomalias nasais
Prevalência:
- 1 em 10.000 nascimentos.
Diagnóstico ultrassonográfico:
- Há um espectro de anormalidades nasais de linha média associadas a holoprosencefalia, incluindo: arrinia ( completa ausência de estruturas nasais), probosis ( apêndice de tecido mole que se projeta diretamente abaixo da testa) e narina única ( usualmente central).
Anormalidades associadas:
- Anomalias cromossômicas, principalmente trissomia 13, são vistas em 40% dos casos.
- Holoprosencefalia em todos os casos. Síndromes genéticas em 20% dos casos.
Investigação:
- Ultrassonografia detalhada.
- Teste invasivo para cariótipo e array.
Seguimento:
- Se gravidez continuar, acompanhamento rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Reservado devido à associação com holoprosencefalia e anomalias cromossômicas.
Recorrência:
- Isolada: 6%.
- Parte da trissomia 13: 1%.
- Parte de síndromes genéticas: 25-50%.
Pescoço
Tópicos nessa seção
- Teratoma cervical
- Higroma cístico
- Bócio tireoidiano
Teratoma cervical
Prevalência:
- 1 em 50.000 nascimentos. • 5% dos teratomas fetais.
Diagnóstico ultrassonográfico:
- Massa sólida vascularizada com componentes císticos, localizada anteriormente ou antero-lateralmente ao pescoço fetal. O tumor cresce rapidamente (especialmente >26 semanas devido aos estrogênios maternos) e pode se estender internamente levando à hiperextensão do pescoço e polidramnia.
- Calcificações são vistas em 50% dos casos.
Anormalidades associadas:
- A incidência de defeitos cromossômicos e síndromes genéticas não é aumentada.
- Tumores grandes podem resultar em anemia fetal e trombocitopenia ( devido a sequestro de hemácias e plaquetas pelo tumor), insuficiência fetal, hidropsia e placentomegalia ( devido à circulação hiperdinâmica como resultado de shunt arteriovenoso), polidramnia ( devido a transudação direta para cavidade amniótica e poliúria fetal secundária à circulação hiperdinâmica) e síndrome espelho materna ( sobrecarga hídrica e pré-eclâmpsia).
Investigação:
- Ultrassonografia detalhada, incluindo ecocardiograma para avaliação de função cardíaca e medida do pico de velocidade sistólica da artéria cerebral média (PVS ACM) para diagnóstico de anemia fetal.
- RMN com 32 semanas para avaliar relação com estruturas adjacentes
Seguimento:
- Ultrassom a cada 2-3 semanas para monitorar crescimento do tumor, função cardíaca, PVS ACM e volume de líquido amniótico.
- Coagulação a laser de vasos tumorais guiada por ultrassom, transfusão intrauterina e amniodrenagem podem se tornar necessários.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas. Mais precocemente se houver evidência de restrição de crescimento, hipóxia fetal ou hidropsia.
- Via: cesariana com procedimento EXIT.
Prognóstico:
- Morte fetal ou neonatal ( devido à obstrução de via aérea) em 80% dos casos.
- Sobrevivência após cirurgia é >80%, entretanto, dissecção cervical extensa e múltiplos procedimentos adicionais são necessários para atingir ressecção completa do tumor com resultados aceitáveis funcional e esteticamente.
Recorrência:
- Não há risco aumentado.
Higroma cístico
Prevalência:
- 1 em 800 gestações. • 1 em 8.000 nascidos vivos.
Diagnóstico ultrassonográfico:
- Estruturas císticas bilaterais simétricas localizadas na região occípito-cervical do pescoço fetal. São diferenciados de edema nucal pela presença do ligamento nucal (septo mediano).
- O higroma cístico é causado por defeito na formação do sistema linfático cervical. É a forma mais comum de linfangioma (75% são localizados no pescoço, 20% na região axilar e 5 % na parede torácica, abdominal e extremidades).
Anormalidades associadas:
- Anomalias cromossômicas, principalmente síndrome de Turner, são vistas em 50% dos casos.
- Síndromes genéticas são vistas em aproximadamente 40% dos casos. As mais comuns são Sd. Noonan (autossômica dominante mas >90% são devido a mutações novas; higroma cístico, hipertelorismo, estenose pulmonar, restrição de crescimento), Sd. Pterígio Múltiplo ( autossômica recessiva; higroma cístico, contratura de todas as articulações, microcefalia e micrognatia), Sd Fryns ( autossômica recessiva; anoftalmia, fenda facial, micrognatia, ventriculomegalia, hérnia diafragmática) e Sd. Neu-Laxova ( autossômica recessiva; hipertelorismo, microcefalia, agenesia de corpo caloso, contraturas de membros superiores e inferiores e restrição de crescimento fetal).
- Hidropsia ( além de higroma cístico há edema generalizado, ascite, derrame pleural e pericárdico) ocorre em 60-80% dos casos.
Investigação:
- Ultrassonografia detalhada, incluindo ecocardiograma.
- Teste invasivo para cariótipo e array.
Seguimento:
- Ultrassom a cada 4 semanas para avaliar evolução do higroma e desenvolvimento de hidropsia.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas. Mais precocemente em caso de hidropsia.
- Via: cesariana em caso de hidropsia ou higroma grande impedindo a flexão da cabeça.
Prognóstico:
- Morte fetal: 90%.
- Em 10% dos casos o cariótipo é normal, não há defeitos fetais óbvios e o higroma se resolve durante a gestação. Nesses casos o prognóstico é bom.
Recorrência:
- Isolado ou parte da síndrome de Turner: não há risco aumentado.
- Parte de síndromes autossômicas recessivas: 25%.
Bócio tireoidiano
Prevalência:
- 1 em 5.000 nascimentos.
Diagnóstico ultrassonográfico:
- Massa cervical anterior ecogênica de tamanho variável. A cabeça fetal pode estar hiperestendida e polidramnia é comum devido à obstrução mecânica do esôfago.
- A maioria dos casos de bócio tireoidiano são consequência de hipotireoidismo fetal por ação transplacentária de drogas anti-tireoidianas usadas para tratamento de hipertireoidismo materno. Uma causa menos comum de bócio hipotiróideo é dishormonogenese congênita devido a defeitos em genes envolvidos na via de produção do hormônio tireoidiano. Fetos com hipotireoidismo podem ter problemas de crescimento e bradicardia.
- Bócio hipertireóideo é raro e causado por ação transplacentárias de imunoglobulinas tireoidianas estimulantes em mãe com doença de Graves diagnosticada recentemente. O feto pode apresentar problemas de crescimento, taquicardia, insuficiência cardíaca e diminuição da movimentação.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada.
- Na maioria dos casos, avaliação da condição materna ajuda a decidir se a causa é hipo ou hipertireoidismo fetal. Em casos duvidosos, cordocentese e medida de hormônios tireoidianos e TSH no sangue fetal, podem distinguir entre hipotireoidismo ( hormônios baixos e TSH alto) devido à drogas anti-tireoidianas ou dishormonogenese congênita e hipertireoidismo ( hormônios altos e TSH baixo) devido a imunoglobulinas tireoidianas estimulantes.
Terapia:
- Bócio hipotireóideo: redução ou até mesmo interrupção da medicação anti-tireoidiana materna com o objetivo de manter os níveis séricos de tiroxina materna no limite superior da normalidade. A segunda linha de tratamento é injeção intra-amniótica de levotiroxina (100?g/kg) cada 1-2 semanas até parto a termo. O bócio, em geral, diminui em tamanho após alguns dias do início do tratamento. Injeções subsequentes são dadas dependendo de evidências sonográficas de aumento da glândula ou medidas seriadas dos níveis hormonais no líquido amniótico ou sangue fetal.
- Bócio hipertireóideo: administração de drogas anti-tireoidianas à mãe. O bócio, em geral, diminui de tamanho em alguns dias, mas se não diminuir, medida dos níveis hormonais no sangue fetal podem ser necessárias e as doses de drogas anti-tireoidianas ajustadas conforme necessário.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar crescimento fetal, tamanho do tumor, frequência cardíaca fetal, volume de líquido amniótico e comprimento de colo.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: cesariana e procedimento EXIT caso haja hiperextensão do pescoço e polidramnia.
Prognóstico:
- Bom.
- Hipotireoidismo congênito não tratado é associado com retardo do neurodesenvolvimento.
Recorrência:
- Dishormonogenese tireoidiana é autossômica recessiva: 25%
- O restante dos caso: não há risco aumentado.
Tórax
Tópicos nessa seção
- Atresia Brônquica
- Cisto Broncogênico
- Síndrome de Obstrução Congênita de Via Aérea Alta
- Malformação Pulmonar Congênita
- Hérnia Diafragmática
- Hipoplasia-Agenesia Pulmonar
- Derrame Pleural
- Sequestro Pulmonar
Atresia brônquica
Prevalência:
- 1 em 100.000 nascimentos.
Diagnóstico ultrassonográfico:
- Pulmão aumentado, hiperecogênico com árvore brônquica dilatada levando a desvio contralateral do mediastino.
- A presença de ascite ( resultado de compressão venosa central) é fator de pior prognóstico.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada.
Seguimento:
- Ultrassom a cada 4 semanas para avaliar evolução da condição.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: cesariana e devido à necessidade de ressuscitação pós-natal imediata e intubação.
Prognóstico:
- Alta taxa de mortalidade.
Recorrência:
- Não há risco aumentado.
Cisto brocogênico
Prevalência:
- 1 em 50.000 nascimentos.
Diagnóstico ultrassonográfico:
- Cisto único em posição central sem sinais de compressão e sem alteração da ecogenicidade do pulmão adjacente.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada.
Seguimento:
- Ultrassom a cada 4 semanas para avaliar evolução da lesão.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: se houver evidência de obstrução brônquica, parto deve ser cesáreo com procedimento EXIT.
Prognóstico:
- Os cistos são, em geral, assintomáticos, exceto em caso de obstrução traqueobrônquica.
Recorrência:
- Não há risco aumentado.
Síndrome de Obstrução Congênita da Via Aérea Alta
Prevalência:
- 1 em 50.000 nascimentos.
Diagnóstico ultrassonográfico:
- Agenesia ou estenose de segmento da via aérea superior na altura da traqueia ou laringe. Em alguns casos, a condição resulta da presença de cistos laríngeos ou membrana laríngea.
- Achados ultrassonográficos se tornam evidentes ? 16 semanas. Os pulmões estão muito aumentados e hiperecogênico levando à compressão cardíaca e desenvolvimento de ascite. A árvore brônquica é dilatada e o diafragma invertido.
Anormalidades associadas:
- A incidência de anomalias cromossômicas não é aumentada.
- Síndromes genéticas são vistas em > 50% dos casos. A mais comum é Sd. Fraser ( autossômica recessiva; microftalmia, fenda facial, Atresia traqueal, agenesia renal bilateral, defeitos cardíacos, sindactilia ou polidactilia.
Investigação:
- Ultrassonografia detalhada.
- RMN pode ajudar a identificar o local e tipo de obstrução.
Seguimento:
- Se a gravidez continuar, ultrassom seriado deve ser realizado para definir melhor momento para parto a depender da evolução da hidropsia.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: cesariana com procedimento EXIT e traqueostomia.
Prognóstico:
- Condição altamente letal com a maioria dos casos evoluindo a óbito no período neonatal.
- Há alguns poucos casos de resolução espontânea intraútero, ou por doilatação de segmento estenótico entre laringe e traqueia, ou por desenvolvimento de fístula traqueoesofágica com drenagem do fluido brônquico para o esôfago.
Recorrência:
- Isolada: não há risco aumentado.
- Síndrome de Fraser: 25%.
Malformação Pulmonar Congênita
Prevalência:
- 1 em 4.000 nascimentos.
Diagnóstico ultrassonográfico:
- Tumor hiperecogênico no tórax fetal, usualmente visto ? 16 semanas. Dividido em sólido ou microcístico, macrocístico com 1 ou mais cistos grandes (>2cm) e misto com áreas sólidas entremeadas a áreas contendo múltiplos cistos < 2cm.
- A lesão é unilateral em >95% dos casos e usualmente envolve 1 lobo ou segmento pulmonar.
- Hidropsia é vista em <10% dos casos.
- Polidramnia, devido à compressão e obstrução esofagiana, pode ser vista ? 26 semanas .
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
- Defeitos em outros sistemas, principalmente cardíaco, renal e fístulas traqueoesofágicas são vistos em 10% dos casos.
Terapia Fetal:
- Shunt tóraco-amniótico pode ser útil em casos de cistos grandes causando importante desvio do mediastino e/ou ascite.
- Em poucos casos de hidropsia, cirurgia fetal aberta com excisão da lesão foi realizada com resultados relativamente bons.
- Há algumas evidência de que administração materna de corticoide pode causar diminuição da lesão e resolução da hidropsia.
Seguimento:
- Ultrassom a cada 4 semanas para avaliar crescimento fetal, lesão pulmonar e volume de líquido amniótico.
- Durante o início do 3o trimestre, >80% das lesões microcísticas se resolve, mas >80% desses casos a resolução não é real, apenas inabilidade de detecção sonográfica pois os pulmões normais também se tornam ecogênicos. A lesão pode ser detectada no período pós-natal através de RX ou mais precisamente por TC.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas. Mais precocemente se houver evidência de restrição de crescimento, hipóxia fetal ou hidropsia.
- Via: indução de parto vaginal.
Prognóstico:
- Sem hidropsia: sobrevivência >95%.
- Hidropsia: usualmente letal.
Recorrência:
- Não há risco aumentado.
Hérnia diafragmática
Prevalência:
- 1 em 4.000 nascimentos.
Diagnóstico ultrassonográfico:
- Vísceras abdominais herniadas para o tórax através de defeito diafragmático associado a desvio do coração de sua posição normal. •
- Intestinos, estômago e/ou fígado no tórax.
- Esquerda (80%), direita (15%) e póstero-lateral ou retroesternal anterior (5%).
- Polidramnia ? 26 semanas na maioria dos casos.
Anormalidades associadas:
- Defeitos cromossômicos, principalmente trissomia 18 ou 13 e ocasionalmente tetrassomia 12p ou Sd. Pallister-Killian são vistos em 20% dos casos.
- Síndromes genéticas são vistas em 10% dos casos. A mais comum é Sd. Fryns ( autossômica recessiva; anoftalmia, fenda facial, micrognatia, ventriculomegalia, hérnia diafragmática).
- Defeitos em outros sistemas, principalmente craniofacial e cardíaco, são vistos em 20% dos casos.
Investigação:
- Ultrassonografia detalhada, incluindo ecocardiograma.
- Amniocentese para cariótipo ( BVC não é apropriada para Sd. Pallister-Killian) e array.
- Avaliação da gravidade é feita através da medida da razão da área do pulmão contralateral no corte transverso do tórax sobre circunferência cefálica (LHR). A razão é comparada àquela esperada para bebês normais e a doença é classificada em severa se a razão ?25%, moderada entre 26-45% e leve se >45%.
- RMN ultra rápida pode ser útil para avaliação acurada da proporção de fígado dentro do tórax.
Terapia Fetal:
- FETO ( oclusão traqueal endoluminal fetoscópica do inglês fetoscopic endoluminal tracheal occlusion). Isso envolve a inserção endoscópica de balão inflável dentro da traqueia com consequente retenção de líquido produzido pelos pulmões o qual estimula o crescimento pulmonar. O balão é inserido com 26 semanas e removido com 34 semanas.
- Um estudo internacional randomizado está investigando a efetividade do FETO quando comparada à conduta expectante em reduzir a mortalidade e morbidade.
Seguimento:
- Ultrassom a cada 4 semanas para avaliar crescimento pulmonar e volume de líquido amniótico. Amniodrenagem pode ser necessária caso haja polidramnia e encurtamento cervical.
- Risco de morte fetal de cerca de 5%.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas. Mais precocemente se houver evidência de restrição de crescimento ou hipóxia fetal.
- Via: indução de parto vaginal.
Prognóstico:
- Parte da Trissomia 18: letal.
- Isolada: sobreviv6encia é < 15% para doença grave, 50% para moderada e > 90% para forma leve.
- Morbidade nos sobreviventes inclui retardo no desenvolvimento em até 30% ( especialmente os que requerem ECMO), refluxo gastroesofágico em 50% e escoliose em 10% dos casos.
Recorrência:
- Isolada: não há risco aumentado.
- Parte de trissomia: 1%.
- Parte de síndromes: 25%.
Agenesia-hipoplasia pulmonar
Prevalência:
- 1 em 50.000 nascimentos.
Diagnóstico ultrassonográfico:
- Completa ausência ou Hipoplasia de um pulmão, em geral o direito, com grande desvio do mediastino.
Anormalidades associadas:
- A incidência de anomalias cromossômicas não é aumentada.
- A associação de pulmão direito hipoplásico com artéria pulmonar direita hipoplásica e drenagem venosa parcial do pulmão direito para veia cava inferior é conhecida como Síndrome Scimitar.
Investigação:
- Ultrassonografia detalhada, incluindo ecocardiograma.
Seguimento:
- Ultrassom a cada 4 semanas para avaliar evolução da condição.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: indução de parto vaginal.
Prognóstico:
- Prognóstico é pior para agenesia direita do que para esquerda, provavelmente por causa do maior grau de desvio do mediastino. Na síndrome Scimitar, a mortalidade é cerca de 10% devido à hipertensão pulmonar severa.
Recorrência:
- Não há risco aumentado.
Derrame pleural
Prevalência:
- Quilotórax congênito isolado é visto em 1 em 10.000 nascimentos.
Diagnóstico ultrassonográfico:
- Usualmente a apresentação se dá em torno de 26 semanas com polidramnia.
- Área anecóica unilateral (25%) ou bilateral ao redor do pulmão. Subjetivamente classificada em leve, moderada ou severa. Nessa última, se unilateral, há desvio do mediastino.
- Em metade dos casos, o derrame é isolado e na outra metade há hidropsia associada com edema subcutâneo e/ou ascite.
Anormalidades associadas:
- Defeitos cromossômicos, principalmente trissomia 21 e monossomia X, são vistos em 10% dos casos.
- Síndrome de Noonan (autossômica dominante mas >90% são devido a mutações novas; higroma cístico, hipertelorismo, estenose pulmonar, restrição de crescimento), é vista em <5% dos casos de hidrotórax isolado.
- Em caso de hidropsia associada, há um grande espectro de condições genéticas, especialmente se houver outros defeitos.
Investigação:
- Ultrassonografia detalhada, incluindo ecocardiograma.
- Teste invasivo para cariótipo e array.
- Investigação por especialistas, incluindo rastreio infeccioso e avaliação para anemia e desordens metabólicas, podem ser necessárias no caso de hidropsia.
Terapia Fetal:
- Em casos graves de derrame pleural uni ou bilateral a colocação de shunt tóraco-amniótico restaura a anatomia torácica normal e resulta na resolução da hidropsia e polidramnia associadas. Alternativa ao shunt é a pleurodese, injeção de substância esclerosante na cavidade pleural.
Seguimento:
- Ultrassom a cada 2 semanas para avaliar evolução do derrame e hidropsia associada. Nova inserção de shunt pode ser necessária em caso de oclusão ou deslocamento.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: cesariana em caso de hidropsia severa. Em fetos com shunt, esse deve ser clampeado no momento do parto para prevenção de pneumotórax.
Prognóstico:
- Isolado: sobrevivência >90%.
- Derrame com hidropsia: em 50% dos casos a hidropsia é resolvida após colocação de shunt tóraco-amniótico e nesses casos, o prognóstico é bom. Se não houver resolução da hidropsia pode haver síndrome genética ou infecção subjacente e nesses casos o prognóstico é ruim.
Recorrência:
- Isolado ou associado à infecção: não há risco aumentado.
- Parte de trissomia: 1%.
- Parte de síndrome genética: até 25%.
Sequestro pulmonar
Prevalência:
- 1 em 15.000 nascimentos.
Diagnóstico ultrassonográfico:
- Massa hiperecogênica no pulmão, na maior parte das vezes no lobo inferior esquerdo.
- Color Doppler demonstra vaso supridor que é ramo direto da aorta descendente.
- Em 75% dos casos é intralobar, sendo indistinguível da aparência da MPA microcística.
- Em 25% dos casos é extralobar localizada fora do pulmão normal com sua própria pleura visceral; na maioria desses casos, há derrame pleural associado.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
- Defeitos em outros sistemas, principalmente, hérnia diafragmática e anomalias cardíacas ou vertebrais, são vistos em até 50% dos casos com sequestro extralobar.
Terapia Fetal:
- Coagulação a laser do vaso supridor guiada por ultrassom nos casos graves de hidrotórax ou hidropsia.
Seguimento:
- Ultrassom a cada 4 semanas para avaliar crescimento do tumor e hidrotórax ou hidropsia.
- Em 30% dos casos há regressão ou desaparecimento do tumor no 3o trimestre.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: indução de parto vaginal.
Prognóstico:
- Sobrevivência: > 95%.
- Terapia Pós-Natal: remoção endoscópica da massa ou embolização seletiva do vaso supridor.
Recorrência:
- Não há risco aumentado.
Parede abdominal
Tópicos nessa seção
- Extrofia vesical
- Anomalia de Body Stalk
- Extrofia Cloacal
- Onfalocele
- Gastrosquise
Extrofia vesical
Prevalência:
- 1 em 30.000 nascimentos.
- Mais comum em homens do que em mulheres: 2:1.
Diagnóstico ultrassonográfico:
- Massa na parede abdominal em região supra-púbica abaixo da inserção do cordão umbilical que é mais baixa que o normal. •
- Bexiga não visualizada com volume normal de líquido amniótico.
- Ossos púbico muito distantes, em homens, pênis curto e largo, em mulheres, hemiclitóris em cada lado da bexiga.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada.
- Teste invasivo para determinar sexo fetal.
Seguimento:
- Rotineiro.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: indução de parto vaginal.
Prognóstico:
- Sobrevivência é > 95%. Cirurgia visa fechamento da bexiga, continência urinária e reparo de epispádia.
Recorrência:
- Não há risco aumentado.
Anomalia de body stalk
Prevalência:
- 1 em 10.000 gestações.
- Mais comuns em mulheres mais jovens e usuárias de cocaína.
Diagnóstico ultrassonográfico:
- Presença de grande defeito de parede abdominal, escoliose severa e cordão umbilical curto ou ausente. Tipicamente, o fígado está diretamente ligado à placenta sem interposição do cordão umbilical e há grande distorção da coluna.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
- Exencefalia ou encefalocele, fendas faciais e amputações de membros são comuns.
Investigação:
- Ultrassonografia detalhada.
Seguimento:
- Se a gravidez continuar, acompanhamento padrão.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Condição letal intra-útero ou no período neonatal.
Recorrência:
- Não há risco aumentado.
Extrofia cloacal
Prevalência:
- 1 em 300.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Onfalocele baixa, bexiga não visualizada e espinha bífida sacral ( em 50% dos casos) com volume de líquido amniótico normal.
- A anatomia da extrofia cloacal é complexa mas, basicamente há uma Onfalocele baixa na margem superior do defeito, intestino delgado ou grosso herniado no meio de 2 hemi-bexigas bastante separadas, Hipoplasia de cólon e atresia anal, ossos púbicos grandemente separados, pênis bífido em homens, hemiclitóris a cada lado da bexiga em mulheres e espinha bífida.
Anormalidades associadas:
- A incidência de anomalias cromossômicas não é aumentada.
- Outros defeitos, principalmente renal e vertebral, pé torto, artéria umbilical única, genitália ambígua ( em homens, o pênis é dividido e duplicado) são comumente vistos.
Investigação:
- Ultrassonografia detalhada.
- Teste invasivo para determinar sexo fetal.
Seguimento:
- Rotineiro.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: indução de parto vaginal.
Prognóstico:
- Sobrevivência: >90% após reconstrução cirúrgica extensiva da bexiga, intestinos e genitália.
- Ambos homens e mulheres com essa condição são capazes de ter vida normal e ambos os sexos são férteis após cirurgia. Alguma forma de desvio no trata urinário é necessária para todos.
Recorrência:
- Não há risco aumentado.
Onfalocele
Prevalência:
- Apenas alças intestinais no saco: 1 em 100 com 11 semanas, 1 em 800 com 12 semanas, 1 em 2.000 com 13 semanas.
- Fígado no saco: 1 em 3.5000 fetos entre 11 e 13 semanas.
Diagnóstico ultrassonográfico:
- Saco herniário em linha média contendo intestino e/ou fígado com cordão umbilical no seu ápice.
- Onfalocele alta pode conter o coração ( Pentalogia de Cantrell).
- Onfalocele baixa pode estar associada a anormalidade cloacal e espinha bífida ( Complexo OEIS).
- Onfalocele contendo apenas intestino com 11-13 semanas tem resolução espontânea em 90% dos casos.
Anormalidades associadas:
- Defeitos cromossômicos, principalmente trissomia 18 ou 13, são vistos em 30-50% dos casos.
- Síndromes genéticas, principalmente Sd. Beckwith-Wiedemann, são vistas em 10% dos casos.
- Defeitos em outros sistemas, principalmente cardíaco, são vistos em 30-50% dos casos.
Investigação:
- Ultrassonografia detalhada, incluindo ecocardiograma.
- Teste invasivo para cariótipo e teste molecular para Sd. Beckwith-Wiedemann.
Seguimento:
- Ultrassom a cada 4 semanas para avaliar crescimento e líquido amniótico. É melhor monitorar o crescimento estimando-se o peso fetal pela fórmula Sieme que usa o diâmetros biparietal e occipitofrontal e comprimento do fêmur, ao invés de utilizar fórmulas que incluem a circunferência abdominal.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas. Mais precocemente se houver evidência de restrição de crescimento ou hipóxia fetal.
- Via: indução de parto vaginal. Cesariana é reservada para casos com indicação obstétrica como apresentação pélvica e onfaloceles gigantes ( saco herniário contendo >75% do fígado) para evitar rotura e hemorragia.
Prognóstico:
- Isolada pequena/moderada: sobrevivência >90%.
- Isolada gigante: sobrevivência 80%.
- Outros defeitos: depende do defeito, p.ex. trissomia 18 é letal.
Recorrência:
- Isolada: não há risco aumentado.
- Parte de trissomia: 1%. • Parte da Sd. Beckwith-Wiedemann: até 50%.
Gastrosquise
Prevalência:
- 1 em 3.000 nascimentos.
- Mais comuns em mulheres mais jovens e usuárias de cocaína.
Diagnóstico ultrassonográfico:
- Defeito paraumbilical de parede abdominal, usualmente à direita, com evisceração intestinal, que flutua livremente no líquido amniótico e cordão umbilical normalmente inserido.
Anormalidades Associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não está aumentada.
Investigação:
- Ultrassonografia detalhada.
Complicações associadas:
- Atresia intestinal ou obstrução secundária a volvo e/ou isquemia no orifício herniário em 10-30%.
- Restrição de crescimento fetal em 30-60% dos casos.
- Parto prematuro espontâneo em cerca de 30%.
- Morte fetal em 2-4% dos casos.
Seguimento:
- Ultrassom a cada 4 semanas para avaliar crescimento, líquido amniótico, oxigenação fetal ( PI-AU, PI-ACM e PI_DV) e dilatação de alça intra-abdominal .
- Em fetos com defeitos de parede abdominal é melhor monitorar crescimento estimando-se o peso fetal pela fórmula Sieme que usa o diâmetros biparietal e occipitofrontal e comprimento do fêmur, ao invés de utilizar fórmulas que incluem a circunferência abdominal.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas. Mais precocemente se houver evidência de restrição de crescimento, hipóxia fetal ou dilatação de alça intra-abdominal (>20mm).
- Via: indução de parto vaginal. Não há evidência que cesariana seja benéfica.
Prognóstico:
- Sobrevivência: >90%.
- Principal causa de morte: síndrome de intestino curto.
Recorrência:
- 3%
Trato gastrointestinal
Tópicos nessa seção
- Cistos Abdominais
- Atresia Anorretal
- Atresia Duodenal
- Atresia Esofagiana
- Calcificações Hepáticas
- Doença de Hirschsprung
- Tumores Hepáticos
- Peritonite Meconial
- Vesícula Biliar não visível
- Obstrução de Intestino Delgado
Cistos abdominais
Massas císticas abdominais são achados frequentes em exames de ultrassom. Anomalias de trato urinário e dilatação intestinal são as explicações mais comuns, apesar de que estruturas císticas podem surgir da árvore biliar, ovários, mesentério e útero. O diagnóstico correto dessas anormalidades pode não ser possível através da ultrassonografia, mas o diagnóstico mais provável é, em geral, sugerido pela posição do cisto e sua relação com outras estruturas e normalidade de outros órgãos.
Cisto de Colédoco:
Cistos de colédoco representam dilatação cística do ducto biliar comum. Diagnóstico pré-natal pode ser feito através de ultrassonografia pela demonstração de cisto na região superior direita do abdome fetal. Há comunicação entre ducto biliar comum e o cisto. Ausência de polidramnia e peristalse ajudam a diferenciar de desordens intestinais. No período pós-natal, diagnóstico precoce e remoção do cisto podem evitar desenvolvimento de cirrose biliar, hipertensão porta, formação de cálculos ou adenocarcinoma. A mortalidade cirúrgica é cerca de 10%.
Cistos mesentérico ou omental:
Esses cistos podem representar obstrução na drenagem linfática. O conteúdo pode ser seroso, quiloso ou hemorrágico. O diagnóstico pré-natal é sugerido pelo achado de lesão cística, unilocular ou multiseptada, usualmente na linha média de tamanho variável; em caso de hemorragia pode haver aparência sólida. Aspiração antenatal pode ser considerada em caso de cistos enormes que resultam em compressão torácica. Manejo pós-natal é conservador e cirurgia é reservada aos casos com sintomas de obstrução intestinal ou dor abdominal aguda após torção ou hemorragia intra-cística. A excisão completa dos cistos pode não ser possível devido à proximidade de vasos sanguíneos de maior calibre e em até 20% dos casos há recorrência pós-cirúrgica. Apesar de transformação maligna em cistos mesentéricos ter sido descrita, esse fato é raro.
Cistos Hepáticos:
Cistos hepáticos são tipicamente localizados no lobo direito do fígado. Eles são raros e resultam da obstrução do sistema biliar hepático. São cistos uniloculares intra-hepáticos e, em geral, assintomáticos, apesar de raramente complicarem com infecções e hemorragias. Cistos hepáticos são vistos em 30% dos adultos com doença renal policística.
Cistos de Duplicação Intestinal:
Esses são bem raros e podem ser localizados ao longo de toda extensão do trato gastrointestinal. À ultrassonografia, aparecem como como estruturas tubulares ou císticas de tamanho variável. Podem ser isolados ou associados a outras malformações gastrointestinais. O diagnóstico pode ser facilitado pela espessura da parede muscular do cisto e presença de peristalse. Remoção cirúrgica é realizada no período pós-natal.
Atresia anorretal
Prevalência:
- 1 em 5.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Sobredistensão do reto e cólon sigmoide no terceiro trimestre.
- Líquido amniótico normal.
- Ocasionalmente, calcificações intraluminais ( mecônio) podem ser vistas.
Anormalidades associadas:
- Defeitos cromossômicos, principalmente trissomia 21 e 18 são vistas em 3-4% dos casos.
- Outros defeitos, principalmente malformações urogenitais, anomalias vertebrais e de SNC são vistos em até 70% dos casos.
- Síndromes não genéticas: Associação VACTERL (esporádica; defeitos vertebrais e de septo ventricular, atresia anal, fístula traqueoesofágica, anomalias renais, displasia radial e artéria umbilical única), Síndrome de Regressão Caudal ( esporádica; agenesia ou Hipoplasia sacral, corpos vertebrais hipoplásicos, atresia anal), Complexo OEIS (esporádico; onfalocele, extrofia cloacal, anus imperfurado e defeitos de coluna).
Investigação:
- Ultrassonografia detalhada.
Seguimento:
- Rotineiro
Parto:
- Local: Hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: indução de parto vaginal.
Prognóstico:
- Em caso isolados, a sobrevivência em geral é boa.
- Em casos com múltiplas anomalias o prognóstico é ruim.
Recorrência:
- Não há risco aumentado.
- Parte de trissomia: 1%.
Atresia duodenal
Prevalência:
- 1 em 5.000 nascidos.
Diagnóstico Ultrassonográfico:
- Sinal da dupla bolha como resultado de aumento do estômago e porção inicial do duodeno. Usualmente visto > 24 semanas.
- Polidramnia > 24 semanas em 50% dos caso.
Anormalidades associadas:
- Defeitos cromossômicos, principalmente trissomia 21, são vistos em 30% dos casos.
- Outros defeitos, principalmente cardíacos, renais, vertebrais, são vistos em 10-20% dos casos.
Investigação:
- Ultrassonografia detalhada, incluindo ecocardiograma.
Seguimento:
- Ultrassom a cada 2-3 semanas para avaliar crescimento e volume de líquido amniótico. Amniodrenagem pode ser necessária em caso de polidramnia e encurtamento de colo.
Parto:
- Local: Hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: indução de parto vaginal.
Prognóstico:
- Taxa de sobrevivência: >95%.
Recorrência:
- Isolada: não há risco aumentado.
- Parte da trissomia 21: 1%
Atresia esofagiana
Prevalência:
- 1 em 3.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Estômago pequeno ou “ausente” na presença de polidramnia > 25 semanas.
- Atresia esofagiana pode ser suspeitada no período pré-natal em apenas 40% dos casos, pois, caso haja fístula traqueoesofágica associada ( vista em >80% dos casos), o estômago parece normal.
Anormalidades associadas:
- Defeitos cromossômicos: trissomia 18 vista em 20% dos casos e trissomia 21 em 1%.
- Outros defeitos, principalmente cardíacos vistos em 50% dos casos.
- Fístula traqueoesofágica pode ser vista como parte da Associação VACTERL (esporádica; defeitos vertebrais e de septo ventricular, atresia anal, fístula traqueoesofágica, anomalias renais, displasia radial e artéria umbilical única).
Investigação:
- Ultrassonografia detalhada, incluindo ecocardiograma.
Seguimento:
- Ultrassom a cada 2-3 semanas para avaliar crescimento e volume de líquido amniótico. Amniodrenagem pode ser necessária em caso de polidramnia e encurtamento de colo.
Parto:
- Local: Hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: indução de parto vaginal.
Prognóstico:
- Sobrevivência é determinada primariamente pela idade gestacional no momento do parto e presença de outras anomalias. Para bebês com fístula traqueoesofágica isolada, nascidos após 32 semanas sem pneumonite por aspiração, sobrevivência pós-operatória é >95%.
Recorrência:
- Isolada: não há aumento no risco.
- Parte de trissomia: 1%.
Calcificações hepáticas
Prevalência:
- 1 em 2.000 fetos com 20 semanas.
Diagnóstico Ultrassonográfico:
- Presença de focos ecogênicos únicos ou múltiplos (1-2mm de diâmetro) dentro do parênquima hepático em sua cápsula.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
- Calcificações no parênquima podem ocorrer devido a infecções intrauterinas.
Investigação:
- Ultrassonografia detalhada.
- TORCH para rastreio de infecção fetal.
Seguimento:
- Em casos isolados, rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Bom.
Recorrência:
- Não há aumento do risco.
Doença de Hirschsprung
Prevalência:
- 1 em 3.000 nascimentos.
Diagnóstico Ultrassonográfico:
- A condição é caracterizada por ausência congênita de gânglios nervosos parassimpáticos intramurais em um segmento do cólon. O segmento agangliônico não é capaz de transmitir a peristalse, logo há acúmulo de mecônio causando dilatação do lúmen intestinal. A aparência ultrassonográfica é semelhante àquela da atresia anorretal quando o segmento afetado é o cólon ou reto.
- Polidramnia e dilatação de alças intestinais são presentes em caso de envolvimento de intestino delgado; nesse caso, não é diferente de outros casos de obstrução.
Anormalidades associadas:
- Anomalias cromossômicas, principalmente trissomia 21, são vistas em 5% dos casos.
Investigação:
- Ultrassonografia detalhada.
- Amniocentese para cariótipo ou análise de cfDNA no sangue materno.
Seguimento:
- Ultrassom a cada 2-3 semanas para avaliar evolução da condição e volume de líquido amniótico. Amniodrenagem pode ser necessária em caso de polidramnia e encurtamento de colo.
Parto:
- Local: Hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: indução de parto vaginal.
Prognóstico:
- Cirurgia pós-natal visa remoção do segmento afetado, podendo ser procedimento em 2 estágios com colostomia temporária. Mortalidade neonatal é aproximadamente 20%.
Recorrência:
- Isolada: Não há risco aumentado.
- Parte da trissomia 21: 1%.
Tumores hepáticos
Prevalência:
- 1 em 100.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Os mais comuns são hemangioma, hamartoma mesenquimal e hepatoblastoma.
- Podem ser císticos, ecogênicos, mistos sólidos e císticos ou podem apenas se apresentar com hepatomegalia.
- Hemangiomas e hepatoblastomas podem ser associados a sinais de falência cardíaca de alto débito, hidropsia e polidramnia.
- Estudos com color Doppler podem distinguir hemangioma de outros tumores.
Anormalidades associadas:
- Anomalias cromossômicas: não há risco aumentado.
- Hepatoblastoma pode ser associado à Síndrome de Beckwith-Wiedemann com sinais de organomegalia e macroglossia.
Investigação:
- Ultrassonografia detalhada, incluindo ecocardiograma.
- Doppler para determinar a anatomia vascular do fígado.
- RMN fetal para determinar a extensão do tumor e sua relação com estruturas intra-hepáticas e órgãos adjacentes.
Seguimento:
- Ultrassom a cada 2-3 semanas para avaliar tamanho do tumor e desenvolvimento de falência cardíaca e polidramnia.
Terapia Fetal:
- Em caso de hemangiomas grandes, associados a falência cardíaca de alto débito, administração materna de corticoide pode fazer cessar o crescimento do tumor.
Parto:
- Local: Hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: cesariana para tumores grandes vascularizados para evitar rotura no momento do parto.
Prognóstico:
- Hemangiomas: sobrevivência de cerca de 80% após tratamento com esteroides. Entretanto, ocasionalmente eles são associados a shunts arteriovenosos , falência cardíaca congestiva e hidropsia, resultando em morte intrauterina ou neonatal. Após a infância, os tumores regridem espontaneamente.
- Hamartoma mesenquimal: ressecção cirúrgica com 70% de sobrevivência.
- Hepatoblastoma: ressecção cirúrgica com 60% de sobrevivência. Antes da ressecção, vários ciclos de quimioterapia podem ser necessários para diminuir o tumor e torná-lo ressecável.
Recorrência:
- Em geral não há risco aumentado.
- No caso de hepatoblastoma associado à Sd. Beckwith Wiedemann (autossômica dominante) e à polipose adenomatosa familiar ( condição autossômica dominante que predispõe a adenomas e carcinomas colônicos) o risco é aumentado.
Mais informações:
Peritonite meconial
Prevalência:
- 1 em 3.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Presença de áreas hiperecogênicas intra-abdominais ( calcificações peritoneais). Resultado de perfuração intrauterina de alças que leva a peritonite química estéril local.
- Além disso: dilatação de alças intestinais, ascite e pseudocisto meconial.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
- Risco de fibrose cística: 75%.
Investigação:
- Ultrassonografia detalhada.
- Amniocentese: estudo de DNA para fibrose cística caso ambos os pais sejam carreadores.
- TORCH para rastreio de infecção fetal.
Seguimento:
- Ultrassom a cada 4 semanas para avaliar evolução da condição.
Parto:
- Local: Hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: indução de parto vaginal.
Prognóstico:
- Em caso de peritonite simples, o desfecho é bom e intervenção cirúrgica não é necessária.
- Em caso de peritonite complexa ( associada à dilatação intestinal, ascite) o prognóstico é ruim e mortalidade neonatal é > 50%.
Recorrência:
- Isolada: não há risco aumentado.
Vesícula biliar não visível
Prevalência:
- 1 em 1.000 fetos com 20 semanas.
Diagnóstico Ultrassonográfico:
- Vesícula biliar pequena ou ausente.
- Na maioria dos casos, esse é achado transitório (75%), mas alguns são devido à agenesia isolada (15%), fibrose cística (10%) e raramente, atresia biliar (3%).
Anormalidades associadas:
- Em cerca de 25% dos casos de atresia biliar há associação com síndrome de heterotaxia que inclui situs inversus, poliesplenia, má rotação intestinal e atresia e defeitos cardíacos.
Investigação:
- Ultrassonografia detalhada.
- Amniocentese: estudo de DNA para fibrose cística caso ambos os pais sejam carreadores.
- Dados limitados sugerem que na atresia biliar há níveis muito baixos de enzimas digestivas no líquido amniótico <24 semanas.
Seguimento:
- Rotineiro
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Atresia biliar é condição séria que requer transplante hepático.
- Agenesia biliar é condição benigna.
Recorrência:
- Não há risco aumentado.
Obstrução de intestino delgado
Prevalência:
- 1 em 5.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Várias alças cheias de líquido no abdome >7cm de diâmetro presentes com >25 semanas.
- Distensão abdominal com peristalse ativa.
- Se perfuração intestinal ocorrer, ascite transitória, peritonite meconial e pseudocistos meconiais podem surgir.
- Polidramnia > 25 semanas, especialmente em obstruções proximais.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
- Outras anomalias intestinais: má rotação, Gastrosquise, duplicação e íleo meconial.
- 10% de risco de fibrose cística ( até 90% caso associada a peritonite meconial).
Investigação:
- Ultrassonografia detalhada.
- Amniocentese: estudo de DNA para fibrose cística caso ambos os pais sejam carreadores.
Seguimento:
- Ultrassom a cada 2-3 semanas para avaliar crescimento e volume de líquido amniótico. Amniodrenagem pode ser necessária caso haja polidramnia e encurtamento de colo.
Parto:
- Local: Hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: indução de parto vaginal.
Prognóstico:
- O prognóstico é relacionado com a idade gestacional no momento do parto, presença de anomalias associadas e local da obstrução. Nos nascidos após 32 semanas com obstrução isolada que requer ressecção de apenas curto segmento intestinal, sobrevivência é >95%. Perda de grandes segmentos de intestino pode levar a síndrome do intestino curto que é condição letal.
Recorrência:
- Isolada: não há risco aumentado.
Trato urinário
Tópicos nessa seção
- Rim em Ferradura
- Hidronefrose
- Tumores Renais
- Rins Multicísticos
- Rim Pélvico
- Doença Renal Policística Autossômica Dominante
- Doença Renal Policística Autossômica Recessiva
- Agenesia Renal
- Obstrução da Junção Uretero-Pélvica
- Obstrução Uretral
- Obstrução da Junção Vésico-Ureteral.
Rim em ferradura
Prevalência:
- 1 em 400 nascimentos.
- Mais comum em fetos masculinos.
Diagnóstico Ultrassonográfico:
- Fusão dos polos inferiores de ambos os rins anteriormente à aorta descendente.
- Rim em ferradura é melhor demonstrado nos cortes coronal e transverso em que tecido renal é visto cruzando a linha média. • Bexiga e volume de líquido amniótico normais.
Anormalidades associadas:
- Anomalias cromossômicas: rim em ferradura é visto em 30% dos casos de Sd. Turner e 20% de trissomia 18.
- Síndromes associadas são vistas em 15% dos casos. A mais comum é a Síndrome de Regressão Caudal (esporádica; agenesia ou hipoplasia sacral, corpos vertebrais hipoplásicos e atresia anal).
- Outros defeitos incluem Hidronefrose e anomalias genitais.
- Defeitos extra-renais, principalmente SNC, cardíacos ou esqueléticos, são vistos em 30% dos casos.
Investigação:
- Ultrassonografia detalhada.
Seguimento:
- Ultrassom a cada 4 semanas para detectar possível desenvolvimento tardio de hidronefrose.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Em formas isoladas, o prognóstico é bom. Acompanhamento pós-natal é recomendado pelo risco significativo de infecção, hidronefrose e nefrolitíase (70%).
Recorrência:
- Isolada ou parte da Sd. Turner: não há aumento no risco.
- Parte da trissomia 18: 1%.
Hidronefrose
Prevalência:
- 1 em 500 nascimentos.
Diagnóstico Ultrassonográfico:
- Dilatação do sistema coletor renal observada no corte transverso padrão do abdome. Ureteres e bexigas normais.
- Dependendo do diâmetro anteroposterior da pelve, a condição é dividida em:
Leve ( apenas pelve renal): 4-7 mm no 2o trimestre; 7-9 mm no 3o.
Moderada ( pelve e cálices): 8-10 mm no 2o trimestre; 10-15 mm no 3o.
Severa (afinamento cortical): >10 mm no 2o trimestre; >15 mm no 3o.
Anormalidades associadas:
- Defeitos cromossômicos: risco baixo em formas isoladas.
- Anormalidades do rim contralateral: rim multicísticos, ectopia, agenesia renal.
- Síndromes associadas são vistas em 5% dos casos.
Investigação:
- Ultrassonografia detalhada (especialmente do rim contralateral).
- Cariótipo deve ser oferecido apenas caso outros marcadores estejam presentes.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar evolução da hidronefrose e avaliar volume de líquido amniótico.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Na maioria dos casos, a condição permanece estável ou se revolve no período neonatal. Em cerca de 20%, pode haver uma obstrução uretero-pélvica ou refluxo vesico-ureteral subjacente o que requer acompanhamento pós-natal e possivelmente cirurgia. Hidronefrose moderada é usualmente progressiva e em mais de 50% dos casos, cirurgia é necessária durante os primeiros 2 anos de vida.
Recorrência:
- Isolada: não há risco aumentado.
Tumores renais
Prevalência:
- 1 em 200.000 nascimentos.
- Nefroma mesoblástico ( hamartoma renal) é o tumor renal mais frequente, enquanto Tumor de Wilms (nefroblastoma) é extremamente raro.
Diagnóstico Ultrassonográfico:
- Em ambos os tumores, há uma massa solitária substituindo a arquitetura normal de todo ou parte do rim.
- Áreas císticas podem aparecer devido à necrose hemorrágica.
- Na maioria dos casos há polidramnia associada.
Anormalidades associadas:
- A incidência de anomalias cromossômicas não é aumentada.
- Nefroblastoma pode ser associada a Síndrome de Beckwith-Wiedemann, com evidência de organomegalia e macroglossia.
Investigação:
- Ultrassonografia detalhada.
Seguimento:
- Ultrassom a cada 2-3 semanas para avaliar tamanho do tumor e desenvolvimento de falência cardíaca e polidramnia.
Parto:
- Local: hospital com UTI neonatal. 20% dos neonatos apresentam hipertensão devido à superprodução de renina pelo rim afetado ou até mesmo pelo normal.
- Via: parto vaginal.
Prognóstico:
- Nefromas mesoblásticos são benignos e a nefrectomia é curativa na maioria dos casos. Em poucos casos há recorrência local ou metástase à distância no primeiro ano de vida.
- Tumor de Wilms é maligno e cerca de 50% são associados a síndromes genéticas ( como Sd. Beckwith-Wiedemann). Tratamento do tumor requer nefrectomia, quimioterapia e às vezes, radioterapia; sobrevivência é > 90%.
Recorrência:
- Nefroma Mesoblástico: não há risco aumentado.
- Em nefroblastoma associado a Sd. Beckwith-Wiedemann (autossômica dominante) o risco é aumentado.
Rins multicísticos
Prevalência:
- 1 em 1.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Os rins são substituídos por múltiplos cistos irregulares de tamanho variável permeados por estroma hiperecogênico.
- Pélvis renais não podem ser visualizadas.
- Pode ser unilateral (80%), bilateral ou segmentar; caso bilateral, há adramnia associada e bexiga “ausente”.
- Anormalidades do rim contralateral (25%): rim duplex, obstrução uretero-pélvica, agenesia, ectopia ou refluxo vesico-ureteral.
Anormalidades associadas:
- Defeitos cromossômicos, principalmente trissomia 18, são vistos em 3% das lesões unilaterais e 15% das bilaterais.
- Síndromes associadas são vistas em 10% dos casos. As mais comuns são: síndrome Branquio-oto-renal (autossômica dominante; cisto branquial – cisto anterolateral no pescoço – rins multicísticos), associação VACTERL (esporádica; defeitos vertebrais e de septo ventricular, atresia anal, fístula traqueoesofágica, anomalias renais, displasia radial e artéria umbilical única), Síndrome de Costela curta e Polidactilia ( autossômica recessiva; membros curtos, tórax hipoplásico, polidactilia, defeitos cardíacos e cerebrais, rins multicísticos), Síndrome de Meckel-Gruber ( autossômica recessiva, polidactilia, rins multicísticos encefalocele occipital).
Investigação:
- Ultrassonografia detalhada.
- Cariótipo pode ser útil nos casos bilaterais ou associados a outras anormalidades para avaliar risco de recorrência.
Seguimento:
- Unilateral: ultrassom a cada 4 semanas para detectar possível desenvolvimento tardio de hidronefrose.
- Bilateral: se a gravidez continuar, acompanhamento rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Bilateral: letal tanto intraútero quanto no período neonatal devido à hipoplasia pulmonar.
- Unilateral: prognóstico normal. No período pós-natal, a maioria dos urologistas adota conduta expectante pois o rim gradualmente encolhe podendo até desaparecer. Pais e a família devem ser submetidos à ultrassonografia para excluir síndrome branquio-oto-renal autossômica dominante.
Recorrência:
- Isolada: 1-2%.
Rim pélvico
Prevalência:
- 1 em 1.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Visualização de rim na pelve acima da bexiga.
- Fossa renal vazia do lado afetado com glândula adrenal preenchendo o espaço.
- Color Doppler pode ser útil na localização do hilo do rim afetado.
- Bexiga e volume de líquido amniótico normais.
Anormalidades associadas:
- A incidência de defeitos cromossômicos e síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada.
Seguimento:
- Ultrassom a cada 4 semanas para detectar possível desenvolvimento tardio de hidronefrose.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Bom prognóstico. Refluxo vésico-ureteral e obstrução da junção uretero-pélvica podem ocorrer.
Recorrência:
- Não há risco aumentado.
Doença renal policística autossômica dominante
Prevalência:
- 1 em 1.000 pessoas são carreadoras do gene mutante.
- Manifestação clínica dessa doença ocorre tipicamente na terceira a quinta década de vida.
Diagnóstico Ultrassonográfico:
- Os rins estão aumentados e hiperecogênicos, mas menores do que na doença autossômica recessiva.
- Pelves renais são visualizadas.
- Bexiga e volume de líquido amniótico são normais.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
- Associação com cistos hepáticos, pancreáticos ou ovarianos.
Investigação:
- Ultrassonografia detalhada.
- Teste genético: a doença é causada por mutações no gene PKD1 (85% dos casos) no cromossomo 16p13.13 e gene PKD2 (15% dos casos) no cromossomo 4q13.23. Diagnóstico pré-natal em famílias afetadas pode ser realizado no primeiro trimestre através de biópsia vilo coriônica.
Seguimento:
- Ultrassom a cada 4 semanas pra avaliar volume de líquido amniótico.
- Ultrassom renal dos pais.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- No aconselhamento de pais com DRPAD deve ser enfatizado que a demonstração ultrassonográfica pré-natal de rins normais não exclui necessariamente a possibilidade do desenvolvimento da doença na vida adulta.
- Diálise e transplante são opções de tratamento quando a patologia se torna sintomática na terceira a quinta décadas de vida.
Recorrência:
- Risco de recorrência: 50%.
Doença renal policística autossômica recessiva
Prevalência:
- 1 em 30.000 nascimentos.
- A doença tem amplo espectro de envolvimento renal e hepático e é subdividida em tipos perinatal, infantil e juvenil dependendo da idade em que ocorre a primeira manifestação clínica.
Diagnóstico Ultrassonográfico:
- Rins aumentados e hiperecogênicos de forma homogênea. Usualmente vistos > 24 semanas.
- Pelves renais não são visualizadas.
- Desenvolvimento gradual de oligodramnia a partir do segundo trimestre.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
- Fibrose cística do fígado: a aparência hepática é normal, apesar da presença de cistos, fibrose portal e proliferação de ductos hepáticos. Quanto mais severa a doença renal, menos severa a fibrose hepática.
Investigação:
- Ultrassonografia detalhada.
- Teste genético: a doença é causada por mutação no gene PKHD1 no cromossomo 6p21. Diagnóstico pré-natal em famílias sob risco pode ser realizado no primeiro trimestre por biópsia vilo coriônicas ( diagnóstico pré-natal confiável em 80% das famílias afetadas).
Seguimento:
- Ultrassom a cada 4 semanas para avaliar crescimento e volume de líquido amniótico.
Parto:
- Local: hospital com UTI neonatal.
- Momento: 38 semanas.
- Via: indução de parto vaginal.
Prognóstico:
- A forma pré-natal é letal ou intraútero ou no período neonatal devido à hipoplasia pulmonar.
- Os tipos infantil e juvenil resultam em falência renal crônica, fibrose hepática e hipertensão porta; muitos casos sobrevivem até adolescência e necessitam de transplante renal.
Recorrência:
- Risco de recorrência: 25%.
Agenesia renal
Prevalência:
- Unilateral: 1 em 2.000 nascimentos.
- Bilateral: 1 em 5.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Unilateral: não visualização de um rim com bexiga e líquido amniótico normais. Color Doppler demonstra artéria renal única. Pode haver hipertrofia compensatória do rim contralateral.
- Bilateral: não visualização dos rins e bexiga em associação a adramnia > 17 semanas. Falência em visualizar artérias renais com color Doppler. Glândulas adrenais assumem formato discoide e se deslocam lateral e inferiormente. Tórax pequeno, hipertrofia cardíaca e pé torto são vistos.
Anormalidades associadas:
- Na maioria dos casos, agenesia renal é esporádica e anomalia isolada.
- Defeitos cromossômicos, principalmente trissomia 18, são vistos em 1-2% dos casos. • Síndromes associadas são vistas em 10% dos casos. As mais comuns são: Sd. Fraser (autossômica recessiva; agenesia renal, atresia laríngea, criptoftalmia, sindactilia), Associação VACTERL (esporádica; defeitos vertebrais e de septo ventricular, atresia anal, fístula traqueoesofágica, anomalias renais, displasia radial e artéria umbilical única), Associação MURCS ( esporádica; útero hipoplásico ou bicorno, agenesia ou displasia renal, anormalidades vertebrais e de costela).
Investigação:
- Ultrassonografia detalhada.
- Cariótipo em casos com anomalias adicionais.
Seguimento:
- Em casos isolados: rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Bilateral é letal, geralmente no período neonatal devido à hipoplasia pulmonar.
- Unilateral tem prognóstico normal. Em alguns pacientes pode haver refluxo vésico-ureteral. Meninas devem ter ultrassonografia pélvica pós-natal a procura de anormalidades em estruturas Müllerianas.
Recorrência:
- Não sindrômico: 3%.
- Em 15% dos casos, um dos pais tem agenesia renal unilateral e nessas famílias o risco de recorrência é aumentado.
Obstrução da junção uretero-pélvica
Prevalência:
- 1 em 2.000 nascimentos.
- Mais comum em homens.
Diagnóstico Ultrassonográfico:
- Dilatação pelvi-caliceal sem dilatação ureteral. O grau de dilatação pelvi-caliceal é variável e, ocasionalmente, urinomas perinéfricos (coleção encapsulada de urina devido à rotura da pelve renal ou cálices ou do ureter) e ascite urinária podem ocorrer.
- Unilateral em 80% dos casos.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
- Anormalidades do rim contralateral: rim multicísticos, ectopia, agenesia renal.
Investigação:
- Ultrassonografia detalhada.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar evolução da hidronefrose e avaliar volume de líquido amniótico.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- A maioria das crianças tem função moderada ou normal podendo ser feita conduta expectante. Pieloplastia é realizada se houver deterioração da função renal.
Recorrência:
- Não há risco aumentado.
Obstrução uretral
Prevalência:
- 1 em 1.500 homens.
Diagnóstico Ultrassonográfico:
- Obstrução uretral pode ser causada por agenesia uretral, persistência da cloaca, estenose uretral ou válvulas de uretra posterior.
- Com válvulas de uretra posterior, em geral, há obstrução incompleta ou intermitente da uretra, resultando em bexiga aumentada e hipertrofiada com vários graus de hidroureter, hidronefrose, e um espectro de displasia renal ( parênquima renal ecogênico com cistos periféricos), oligodramnia e hipoplasia pulmonar.
- Em alguns casos há ascite urinária associada por rotura vesical.
- Quantidade reduzida de líquido amniótico > 17 semanas.
- No primeiro trimestre, a condição se apresenta como megabexiga (diâmetro longitudinal da bexiga ? 7mm). Se o diâmetro vesical é 7-15mm há resolução espontânea do quadro em cerca de 90% dos casos. Se o diâmetro vesical é ?15mm a condição é invariavelmente associada à uropatia obstrutiva progressiva levando a hidronefrose e rins displásicos.
Anormalidades associadas:
- Defeitos cromossômicos, principalmente trissomia 21,18 ou 13, são vistos em cerca de 10% dos casos.
- Outros defeitos, principalmente cardíacos e gastrointestinais são visots em até 40% dos casos.
Investigação:
- Ultrassonografia detalhada.
- Amniocentese para cariótipo ou análise de cfDNA no sangue materno.
Terapia Fetal:
- Descompressão do trato urinário já foi conseguida através da inserção guiada por ultrassom de cateteres suprapúbicos e ablação cistoscópica de válvulas uretrais. Apesar de essas técnicas mostraram ser possível a cirurgia intrauterina, elas não oferecem evidências conclusivas que tais intervenções melhoram a função renal e pulmonar.
Seguimento:
- Se a gravidez continuar, ultrassom a cada 4 semanas para monitorar evolução da condição e volume de líquido amniótico.
- Avaliação pré-natal da função renal depende da combinação de achados ultrassonográficos e análise de urina fetal obtida por urodococentese. Sinais de prognóstico ruim são: – Presença bilateral de rins multicísticos ou hidronefrose severa com córtex ecogênico ou cístico. – Adramnia, sugerindo obstrução uretral completa. – Altos sódio urinário ( mais de 100mg/dL), cálcio (mais de 8mg/dL) e ?2 microglobulina ( mais de 40mg/L).
Parto:
- Local: Hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: indução de parto vaginal.
Prognóstico:
- Severa: alta mortalidade perinatal devido à hipoplasia pulmonar secundária a oligodramnia grave. Mesmo nos que sobrevivem, cerco de 30% desenvolvem falência renal necessitando de diálise e/ou transplante antes de 5 anos de idade.
Recorrência:
- Isolada: não há risco aumentado.
- Parte de trissomia: 1%.
Obstrução da junção uretero-vesical
Prevalência:
- 1 em 10.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Presença de hidroureter e hidronefrose com bexiga normal.
- O ureter dilatado é tortuoso e aparece como coleção de cistos de tamanhos variáveis, localizados entre a pelve renal dilatada e a bexiga.
- A etiologia é diversa incluindo estenose ou atresia ureteral, obstrução vascular, divertículo ou ureterocele. Ureteroceles ( visíveis como áreas circulares pequenas de parede fina, cheias de líquido dentro da bexiga) são usualmente vistas em associação a rim e sistema coletor duplex. O polo superior em geral obstrui e o inferior tem refluxo.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar evolução da hidronefrose e avaliar volume de líquido amniótico.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Intervenção pós-natal é indicada se o refluxo vesico-ureteral é associada a função renal anormal.
Recorrência:
- Não há risco aumentado.
Trato genital
Tópicos nessa seção
- Genitália Ambígua
- Cisto Ovariano
- Seio Urogenital
Genitália ambígua
Prevalência:
- 1 em 5.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Feto feminino: clitoromegalia com lábios normais.
- Feto masculino: micropênis, hipospádias, criptorquidia, escroto bífido.
- Dependendo da causa, a condição é dividida em:
- Hermafroditismo verdadeiro: ambos os tecidos ovariano e testicular estão presentes na mesma gônada. O cariótipo é feminino 46XX, mas há material cromático do cromossomo y.
- Pseudohermafroditismo feminino: meninas virilizadas com cariótipo feminino normal e tecido ovariano na gônada. As causas incluem hiperplasia adrenal congênita (1 em 15.000), ingestão de andrógenos pela mãe e tumores virilizantes maternos.
- Pseudohermafroditismo masculino: meninos subvirilizados com cariótipo masculino normal e tecido testicular. As causas incluem síntese inadequada de testosterona ou presença de defeitos no receptor androgênico.
Anormalidades associadas:
- Anormalidades cromossômicas, principalmente trissomia 13, triploidia e síndrome 13q, são vistas em poucos casos.
- A condição é comumente associada a síndromes genéticas:
- Síndrome Smith-Lemli-Opitz: autossômica recessiva; genitália ambígua, microcefalia, defeitos cardíacos, renais e gastrointestinais, sindactilia e polidactilia.
- Síndrome WAGR: esporádica; tumor de Wilms, aniridia (ausência de íris), malformações geniturinárias, retardo no neurodesenvolvimento.
- Outros defeitos, principalmente fendas faciais e defeitos cardíacos são vistos com frequência.
Investigação:
- Ultrassonografia detalhada.
- Procurar sinais maternos de hiperandrogenismo ( acne, voz grave, hirsutismo durante a gestação) e perguntar sobre ingestão de andrógenos durante o primeiro trimestre, além de história familiar de genitália ambígua.
- Determinar sexo genético através de teste invasivo ou cfDNA no sangue materno.
- Famílias sob risco de hiperplasia adrenal congênita: teste invasivo para análise de DNA.
- Casos suspeitos de Sd. Smith-Lemli-Opitz: Amniocentese e medida de 7-dehidrocolesterol; altos níveis sugerem o diagnóstico.
Seguimento:
- Em famílias com hiperplasia adrenal congênita, administração de dexametasona à grávida desde 6 semanas de gestação pode minimizar o efeito de andrógenos na genitália e no cérebro em desenvolvimento. Se o feto for masculino, o corticoide pode ser descontinuado.
- Ultrassom a cada 4 semanas para monitorar crescimento e evolução da genitália.
Parto:
- Assistência obstétrica de rotina, mas o parto deve ser em centro terciário.
Prognóstico:
- Tratamento de neonato com genitália ambígua deve ser realizado por equipe multidisciplinar, incluindo geneticista, endocrinologista pediátrico e urologista pediátrico. Há controvérsias a respeito da determinação do sexo e necessidade ou não de cirurgia reconstrutiva.
Recorrência:
- Hiperplasia adrenal congênita: 25%.
Cisto ovariano
Prevalência:
- 1 em 2.500 nascimentos.
- Cisto intra-abdominal mais comum nos fetos femininos.
Diagnóstico Ultrassonográfico:
- Cisto unilocular, unilateral, às vezes contendo “cisto filho”, no abdome de feto feminino > 26 semanas.
- Se o cisto sofre torção (40% dos casos) ou hemorragia, a aparência é complexa ou sólida. Rotura pode levar à ascite.
- Cistos ovarianos fetais são sensíveis aos hormônios placentários e são mais comuns em diabéticas ou mães aloimunizadas (Rh) como resultado de hiperplasia placentária.
- Cistos ovarianos grandes (>6 cm em diâmetro) podem causar polidramnia por compressão intestinal.
Anormalidades associadas:
- A maioria dos casos é esporádica e não há associação com anomalias cromossômicas.
- Alguns poucos casos são associados a síndromes genéticas. A mais comum é Síndrome McKusick-Kaufman ( autossômica recessiva; hidrometrocolpos, polidactilia e defeitos cardíacos).
- Outros defeitos, principalmente geniturinários ( agenesia renal, rins policísticos) e gastrointestinais ( atresia esofagiana, atresia duodenal e anus imperfurado) são vistos com frequência.
Investigação:
- Ultrassonografia detalhada.
Seguimento:
- Ultrassom a cada 4 semanas para avaliar evolução do cisto. Se cisto > 6cm, aspiração guiada por ultrassonografia deve ser considerada.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: indução de parto vaginal.
Prognóstico:
- A maioria dos cistos é benigna e se resolve espontaneamente no período neonatal. Cirurgia pode ser necessário caso haja torção.
Recorrência:
- Isolado: não há risco aumentado.
Seio urogenital
Prevalência:
- 1 em 250.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Massa cística pré-sacral na pelve de feto feminino devido a canal comum para trato urinário e genital. A massa representa hidrometrocolpos, vagina distendida e bexiga comprimida e deslocada anteriormente com graus variáveis de obstrução urinária. O conteúdo pode ser claro (urina) ou turvo (hemorragia). Ascite pode resultar do escape de urina através trompa de Falópio para o abdome.
- Quantidade normal de líquido amniótico.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
- Outros defeitos, principalmente do trato geniturinário (útero bicorno, atresia cervical, atresia ou duplicação vaginal, hidronefrose, agenesia renal, rins multicísticos). Trato gastrointestinal ( ânus imperfurado, atresia esofagiana) e do sistema cardiovascular são frequentemente vistos.
Investigação:
- Ultrassonografia detalhada.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar evolução da condição.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: indução de parto vaginal.
Prognóstico:
- Avaliação neonatal do exato quadro anatômico e investigação de anomalias dos tratos genital e urinário. Cirurgia reconstrutora é necessária. A maioria dos paciente é continente e fértil, mas cerca de 50% requer cateterização intermitente.
Recorrência:
- Isolado: não há risco aumentado.
Extremidades
Tópicos nessa seção
- Amputação ou Deficiência de Membro
- Síndrome de Banda Amniótica
- Artrogripose
- Clinodactilia
- Pé Torto
- Mãos Tortas
- Ectrodactilia
- Polidactilia
- Sindactilia
Amputação ou deficiência de membro
Prevalência:
- 1 em 20.000 nascimentos.
Princípios Gerais:
- Em 50% dos casos há deficiência por redução transversa de um antebraço ou mão sem outras anomalias associadas.
- Em 50% dos casos há múltiplas deficiências por redução e em 25% desses há anomalias adicionais dos órgãos internos ou estruturas craniofaciais.
- Amputação da extremidade superior é, em geral, achado isolado, enquanto que, amputação de pernas ou amputações bilaterais de membros são, geralmente, parte de síndrome genética.
- Amputação isolada de uma extremidade pode ocorrer devido à síndrome de banda amniótica, exposição a teratógeno ou acidente vascular. • Se o defeito é unilateral, pode corresponder ao complexo fêmur-fíbula-ulna ou fêmur-tíbia-rádio. O primeiro é não familiar enquanto o segundo, tem forte associação a componente genético.
Focomelia:
- Mãos e pés estão presentes (podem ser normais ou anormais), mas os braços e pernas são ausentes. •
- Três síndromes devem ser consideradas no diagnóstico diferencial de focomelia:
- Síndrome de Roberts: autossômica recessiva; tetrafocomelia, fenda facial bilateral, micrognatia, hipertelorismo e malformações de orelha e nariz.
- Síndrome de Trombocitopenia com Rádio Ausente (TRA): autossômica recessiva; ausência bilateral de rádio associada a trombocitopenia.
- Síndrome de Grebe: desordem autossômica recessiva descrita em tribos indígenas consanguíneas do Brasil. Caracterizada por importante hipomelia dos membros superiores e inferiores, com aumento da severidade dos segmentos proximais para distais. Ao contrário da síndrome de Roberts, os membros inferiores são mais afetados que os superiores.
Fêmur Curto Congênito:
- Classificado em 5 grupos:
- Hipoplasia de Fêmur Simples.
- Fêmur curto com eixo angulado.
- Fêmur Curto com Coxa Vara: esse é o mais comum sendo causado por deformidade do quadril, onde o ângulo entre cabeça e eixo femoral é reduzido a menos do que 120 graus. – Fêmur Proximal Ausente ou Defeituoso.
- Fêmur Ausente ou Rudimentar.
- Um dos dois fêmures pode ser afetada mas o direito é o mais frequentemente envolvido.
- Síndrome de Hipoplasia Femoral e Fácies Não Usuais, a qual é esporádica, consiste em hipoplasia femoral bilateral e defeitos faciais, incluindo nariz curto com ponta larga, filtro longo, micrognatia e fenda palatina.
Síndrome de banda amniótica
Prevalência:
- 1 em 1.200 nascimentos.
Diagnóstico Ultrassonográfico:
- Espectro de características envolvendo extremidades. Região craniofacial e tronco que pode ser isolado ou aparecer de forma combinada. Demonstração da banda não é necessária para o diagnóstico de síndrome de banda amniótica.
- Extremidades: dedos ou porções de membros ausentes, porção distal de braço ou perna edemaciados resultante de bandas amnióticas constritivas.
- Região Craniofacial: fenda facial, microftalmia assimétrica, deformidade nasal grave e encefalocele.
- Tronco: deformidades graves de coluna, defeitos grandes de parede abdominal com herniação intestinal. A manifestação mais extrema é a anomalia de body stalk.
Anormalidades associadas;
- A incidência de anomalias cromossômicas ou síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada.
Seguimento:
- Ultrassom a cada 2-3 semanas para avaliar consequências da banda.
Terapia Fetal:
- Em alguns casos, lise fetoscópica das bandas foi usada em bandas com constrição de cordão umbilical ou risco de amputações de membros.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
Prognóstico:
- Prognóstico e tratamento dependem da natureza da síndrome de banda amniótica e severidade da deformação. Deformidades faciais frequentemente requerem procedimentos de reconstrução extensivos.
Recorrência:
- Não há risco aumentado.
Artrogripose
Prevalência:
- 1 em 3.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Mal posicionamento dos membros e movimentos fetais limitados resultado de contratura em ? 2 articulações.
- O início do quadro de artrogripose varia: de 12 a 30 semanas.
- A condição geralmente está associada à polidramnia (>25 semanas), tórax estreito, micrognatia e edema nucal ( ou translucência nucal aumentada entre 11-13 semanas).
- Contrações fixas anormais de músculos podem ser:
- Regional: apenas os membros superiores ou inferiores afetados. Se a região inferior é afetada, as pernas estão hiperestendidas e cruzadas. Se a região superior é afetada, os bração estão fletidos cada um de um lado do tórax.
- Generalizada: todos os músculos estão afetados como na sequência deformativa de acinesia fetal.
Anormalidades associadas:
- Anomalias cromossômicas: alguns casos têm trissomia 8 ou 18.
- Mais de 150 síndromes genéticas são associadas à artrogripose. As mais comuns são:
- Sequência Deformativa de Acinesia Fetal: grupo de anormalidades de diferentes causas caracterizada por diminuição dos movimentos fetais, contratura de múltiplas articulações, restrição de crescimento, anomalias faciais, hipoplasia pulmonar e, ocasionalmente, hidropsia.
- Síndrome Cérebro-Óculo-Facio-Esquelética: autossômica recessiva; tônus muscular gravemente reduzido, características faciais típicas ( baixa implantação de orelhas, microftalmia, micrognatia) e anormalidades de crânio, membros, coração e rins.
- Síndrome Neu-Laxova: autossômica recessiva; hipertelorismo, microcefalia, agenesia de corpo caloso, contraturas de membros superiores e inferiores e restrição de crescimento fetal.
- Síndrome de Múltiplos Pterígios: autossômica recessiva; higroma cístico, contratura de todas as articulações, microcefalia e micrognatia.
Investigação:
- Ultrassonografia detalhada.
- Teste invasivo para cariótipo e array.
- Histórico médico detalhado em relação à infecção, febre ou hipotermia e exposição a teratógenos com fenitoína e etanol deve ser obtido.
- Distrofia Miotônica e Miastenia Gravis devem ser excluídas.
Seguimento:
- Ultrassom a cada 2-3 semanas para monitorar crescimento, movimentos em todas as articulações e volume de líquido amniótico. Amniodrenagem pode ser necessária em caso de polidramnia e encurtamento do colo.
Parto:
- Local: hospital com UTI neonatal.
- Via: cesariana eletiva devido à possibilidade de complicações resultantes de posição fetal anormal e falta de flexibilidade dos membros.
Prognóstico:
- Se múltiplos órgãos são afetados há alta mortalidade nos primeiros meses de vida.
- Se apenas membros são acometidos, cirurgia e fisioterapia visam atingir a máxima função de cada articulação envolvida.
Recorrência:
- Depende da condição subjacente.
- Artrogripose distal: 50%.
Clinodactilia
Prevalência:
- 1 em 100 nascimentos.
Diagnóstico Ultrassonográfico:
- Desvio medial ou curvatura radial do dedo na altura da articulação interfalangeana distal, afetando, em geral, o quinto dedo.
Anormalidades associadas:
- Anomalias cromossômicas: Clinodactilia bilateral é vista em 60% dos recém-nascidos com trissomia 21.
- A condição é geralmente isolada.
Investigação:
- Ultrassonografia detalhada.
- Teste invasivo e array são recomendados nos caso não isolados.
Seguimento:
- Rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Casos isolados: bom prognóstico. Não há necessidade de cirurgia pós-natal.
Recorrência:
- Isolada familiar: 50%.
- Parte da trissomia 21: 1%.
Pé torto
Prevalência:
- 1 em 1.000 nascimentos.
- Bilateral em 50% dos casos.
Diagnóstico Ultrassonográfico:
- Demonstração de que a sola do pé não é perpendicular aos ossos da perna.
Anormalidades associadas:
- Em > 50% dos casos a condição é isolada.
- Anomalias cromossômicas: achado comum em trissomia 18 e 13.
- Comumente associada a oligodramnia prolongada, anormalidades cerebrais, espinha bífida e desordens esqueléticas e neuromusculares.
- Mais de 250 síndromes genéticas incluem pé torto como um de seus componentes.
Investigação:
- Ultrassonografia detalhada.
- Casos não isolados: teste invasivo e array.
Seguimento:
- Isolado: acompanhamento rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Isolado: bom prognóstico.
- Em 90% dos casos, boa funcionalidade a longo prazo é atingida através de manipulação e aplicação seriada de gesso associada à intervenção cirúrgica mínima.
- Cirurgia é necessária em cerca de 10% dos casos e em um terço, é preciso mais de uma cirurgia.
Recorrência:
- Um filho com pé torto: 3%.
- Um dos pais e um filho com pé torto: 25%.
Mãos tortas
Prevalência:
- 1 em 30.000 nascimentos.
- Bilateral em 50% dos casos.
Diagnóstico Ultrassonográfico:
- Mãos tortas são subdivididas em radial e ulnar:
- Mão Torta Radial: aplasia radial, desvio radial agudo da mão e ausência ou hipoplasia do polegar.
- Mão Torta Ulnar: varia de pequeno desvio da mão para lado ulnar do antebraço até completa ausência da ulna.
Anormalidades associadas:
- Mão torta com desvio ulnar é geralmente anomalia isolada, enquanto a mão torta radial é frequentemente sindrômica:
- Trissomia 18: mão torta radial é achado comum nessa cromossomopatia.
- Síndrome de Trombocitopenia com Rádio Ausente (TRA): aplasia radial, polegares presentes, defeitos cardíacos e micrognatia.
- Síndrome Holt-Oram: Aplasia radial, sindactilia, defeitos cardíacos.
- Síndrome de Roberts: aplasia radial, focomelia, fenda facial, defeitos cardíacos.
- Associação VACTER: aplasia ou hipoplasia radial, defeitos vertebrais e de septo ventricular, atresia anal, fístula traqueoesofágica, anomalias renais e artéria umbilical única.
Investigação:
- Ultrassonografia detalhada.
- Teste invasivo para cariótipo e array.
Seguimento:
- Ultrassom a cada 4 semanas.
- Em famílias com história de síndrome TRA: cordocentese pra diagnóstico de anormalidades hematológicas.
Parto:
- Assistência obstétrica de rotina, porém o parto deve ser realizado em hospital com UTI neonatal.
Prognóstico:
- Depende de condições associadas.
- Tratamento cirúrgico e fisioterapia visam melhorar a mobilidade, força e estabilidade do antebraço e punho.
Recorrência:
- Parte da trissomia 18: 1%.
- Parte de síndromes genéticas: 25-50%.
Ectrodactilia
Prevalência:
- 1 em 20.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Espectro de defeitos de mãos e pés com ausência de dígitos, fenda mediana e fusão dos dígitos restantes resultando em extremidades semelhantes a garras.
Anormalidades associadas:
- Anomalias cromossômicas: vista na trissomia 18.
- Alta incidência em síndromes genéticas. As mais comuns são:
- Síndrome de Roberts: autossômica recessiva; ectrodactilia, focomelia, fenda facial.
- Ectrodactilia-Displasia Ectodérmica (EDE): autossômica dominante; deformidades em todas as 4 extremidades mas mais severa nas mãos e defeitos ectodérmicos ( pele seca, cabelo ralo, defeitos dentários e de ducto lacrimal).
- Síndrome de Nager: autossômica dominante, mas na maioria dos casos mutação nova – de novo – Ectrodactilia, micrognatia, anomalias de orelha externa.
- Malformação de Mãos e Pés Divididos: Ectrodactilia apresentando-se como anomalia de “garras de lagosta”.
Investigação:
- Ultrassonografia detalhada.
- Teste invasivo para cariótipo e array.
Seguimento:
- Rotineiro.
- Consulta com geneticista clínico.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Isolada: bom prognóstico.
- Tratamento cirúrgico e fisioterapia, diminuem a debilidade funcional das mãos.
Recorrência:
- Familiar isolada: 50%.
Polidactilia
Prevalência:
- 1 em 100 nascimentos em negros e 1 em 700 nascimentos em brancos.
- Mais comum em homens que em mulheres: 2:1.
Diagnóstico Ultrassonográfico:
- Mais de 5 dígitos com ou sem com ou sem falange óssea na mão ou no pé.
- Há 2 tipos de polidactilia:
- Pós-axial (mais comum): sexto dedo está do lado ulnar ou fibular, após o quinto dedo.
- Pré-axial (rara): sexto dedo está no lado radial ou tibial, antes do polegar ou hálux.
Anormalidades associadas:
- Anomalias cromossômicas: vista em 75% dos fetos com trissomia 13.
- Na maioria dos casos é achado isolado, com padrão de herança autossômico dominante. Em alguns casos há associação com síndromes genéticas:
- Síndrome de Meckel-Gruber: autossômica recessiva; polidactilia, rins multicísticos, encefalocele occipital.
- Síndrome Bardet-Biedl: autossômica recessiva; polidactilia pós-axial, rins aumentados hiperecogênicos. Pós-natal: obesidade, retinopatia, hipogonadismo, desordens neurológicas.
- Síndrome de Costela curta e Polidactilia: autossômica recessiva; membros curtos, tórax hipoplásico, polidactilia, defeitos cardíacos e cerebrais, rins multicísticos.
- Síndrome Ellis-van Creveld: autossômica recessiva; membros curtos, polidactilia pós-axial, tórax estreito, defeitos cardíacos.
Investigação:
- Ultrassonografia detalhada.
- Teste invasivo e array para casos não isolados.
- Consulta com geneticista clínico.
Seguimento:
- Em casos isolados, rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Casos isolados: bom prognóstico.
Recorrência:
- Familiar isolada: 50%.
- Parte da trissomia 13: 1%.
- Parte de síndromes autossômicas recessivas: 25%.
Sindactilia
Prevalência:
- 1 em 3.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Dois ou mais dígitos ( osso ou tecido mole) estão fundidos.
Anormalidades associadas:
- Anomalias cromossômicas: achado comum em triploidia.
- Na maioria dos casos é achado isolado, mas há associação comum com síndromes genéticas:
- Síndrome de Apert: autossômica dominante; braquisindactilia das mãos e pés, craniossinostose , hipertelorismo e defeitos cardíacos.
- Síndrome de Carpenter: autossômica recessiva; polissindactilia, craniossinostose.
- Síndrome de Fraser: autossômica recessiva; microftalmia, fenda facial, atresia traqueal, agenesia renal bilateral, defeitos cardíacos, sindactilia ou polidactilia
Investigação:
- Ultrassonografia detalhada.
Seguimento:
- Rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Casos isolados: bom prognóstico.
Recorrência:
- Polissindactilia Isolada Familiar: 50%.
- Parte da Síndrome de Apert: 50%.
- Parte da Síndrome de Carpenter e Fraser: 25%.
- Parte de Triploidia: não há risco aumentado.
Esqueleto
Tópicos nessa seção
- Displasia Esquelética
- Acondrogênese
- Acondroplasia
- Distrofia Torácica Asfixiante
- Atelosteogênese
- Displasia Campomélica
- Craniossinostose
- Displasia Distrófica
- Síndrome de Ellis-Van Creveld
- Hipofosfatasia
- Síndrome Jarcho-Levin
- Osteogenese imperfeita
- Síndrome de Costela Curta e Polidactilia
- Displasia Tanatofórica
Displasia esquelética
Prevalência:
- 1 em 4.000 nascimentos.
- 25% são natimortos e 30% morrem no período neonatal.
Abordagem Diagnóstica Pré-Natal:
- Há uma gama de displasias esqueléticas raras, cada uma com risco de recorrência específico, expressão dismórfica e implicações para sobrevivência neonatal e qualidade de vida.
- O achado acidental de displasia esquelética em ultrassonografia de rotina em gravidez sem risco de síndrome específica, necessita de exame sistemático dos membros, cabeça, tórax e coluna para fechar o diagnóstico correto.
Avaliação de ossos longos:
- Encurtamento das extremidades pode envolver o membro inteiro (micromelia), o úmero ou fêmur (rizomelia), o rádio, ulna, tíbia ou fíbula (mesomelia) ou mãos e pés (acromelia). O fêmur é anormalmente curto até mesmo em nanismo mesomélico e, por isso, em rastreio rotineiro de anomalias fetais, o fêmur deve ser medido e comparado subjetivamente a todos os ossos longos. Redução grave de membros como no nanismo tanatofórico e acondrogênese pode ser detectada a partir de 16 semanas, enquanto na acondroplasia, o encurtamento se torna óbvio > 22 semanas.
- Forma anormal. Em algumas condição há uma curvatura pronunciada (p.ex. displasia campomélica, nanismo tanatofórico) e em outras, fraturas e formação de calos podem ser detectadas (p.ex. osteogênese imperfeita, acondrogênese e hipofosfatasia).
- Ecogenicidade reduzida dos ossos devido à hipomineralização é vista em algumas desordens (p.ex. hipofosfatasia, osteogênese imperfeita e acondrogênese). Ausência virtual de ossificação da coluna na acondrogênese pode levar ao diagnóstico errôneo de agenesia vertebral completa. Déficit de mineralização do crânio na hipofosfatasia, pode resultar em diagnóstico equivocado de hidrocefalia.
- Ausência de extremidades, como amelia (completa ausência de extremidades), acheiria (ausência da mão), focomelia (membro de foca) ou aplasia/hipoplasia de rádio e ulna, são comumente herdados como parte de uma síndrome genética (Sd. Holt-Oram, Sd. Trombocitopenia e ausência de rádio). Outra causa de perda focal de membro é a síndrome de banda amniótica.
Avaliação de Mãos e Pés:
- Várias displasia esqueléticas estão associadas com alterações de mãos e pés.
- Polidactilia: Presença de mais de 5 dígitos. Classificada como pós-axial se o dedo extra é na face ulnar ou fibular e pré-axial se é localizado na face radial ou tibial.
- Sindactilia: Fusão de dedos adjacentes através de tecido ósseo ou mole.
- Clinodactilia: desvio de dedo(s).
- Desproporção entre mãos e pés e as outras porções da extremidade também pode ser sinal de displasia esquelética.
Avaliação de Movimentos Fetais:
- Artrogripose e síndrome de múltiplos pterígios são caracterizadas por limitação de flexão ou extensão dos membros.
Avaliação do Tórax Fetal:
- Várias displasias esqueléticas são associadas a tórax pequeno, que leva à hipoplasia pulmonar e morte neonatal. O diagnóstico de tórax pequeno pode ser feito através da razão da circunferência torácica/abdominal ou da razão da circunferência torácica/cefálica. A circunferência torácica é medida no nível do corte de 4 câmaras cardíacas.
Avaliação da Cabeça Fetal:
- Várias displasias esqueléticas são associadas a redução da ossificação craniana. A face também deve ser examinada para diagnóstico de hipertelorismo, micrognatia, lábio superior curto e anormalidades de orelha.
Testes Diagnósticos Complementares à Ultrassonografia:
- Avaliações pré ou pós-natal incluem análise de DNA para grande número de displasias. Algumas podem, agora, ser diagnosticadas por cfDNA no sangue materno. No período pós-natal, radiografias esqueléticas são de grande importância, já que a classificação das displasias é baseada em achados radiográficos.
Acondrogênese
Prevalência:
- 1 em 40.000 nascimentos.
- Segunda mais comum displasia esquelética letal, só atrás da displasia tanatofórica.
Diagnóstico Ultrassonográfico:
- Encurtamento grave dos membros, tórax estreito, tronco curto e cabeça grande.
- Associada a micrognatia, edema nucal e polidramnia.
- Há 2 tipos de acondrogênese:
- Tipo I (20%): autossômica recessiva; há mineralização pobre do crânio e corpos vertebrais assim como fraturas costais.
- Tipo II (80%): esporádica; há mineralização pobre de corpos vertebrais mas não do crânio e não há fraturas costais.
Investigação:
- Ultrassonografia detalhada.
- Acondrogênese ocorre devido a mutações nos genes SLC26A2, COL2A1 e TRP11.
Seguimento:
- Se a gravidez continuar, rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- A condição é letal devido à hipoplasia pulmonar grave.
Recorrência:
- Tipo I: 25%.
- Tipo II: não há risco aumentado.
Acondroplasia
Prevalência:
- 1 em 25.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Membros, mãos e dedos curtos, cabeça grande com bossa frontal e ponde nasal deprimida e escoliose lombar.
- Encurtamento de membros e características faciais típicas se tornam aparentes > 22 semanas.
Investigação:
- Ultrassonografia detalhada.
- Acondroplasia ocorre devido à mutação no gene FGFR3 e tem padrão de herança autossômico dominante. O diagnóstico pode ser atingido através de exame invasivo ou análise de cfDNA no sangue materno.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar crescimento da cabeça fetal.
Parto:
- Assistência obstétrica e parto de rotina.
- Cesariana caso circunferência cefálica > 40cm.
Prognóstico:
- Tipo Homozigoto: letal, devido à severa hipoplasia pulmonar.
- Tipo Heterozigoto: inteligência e expectativa de vida normais. Limitações respiratórias devido ao tórax estreito e desenvolvimento de canal vertebral estenótico ( déficits neurológicos periféricos) podem diminuir a qualidade de vida.
Recorrência:
- Pai/mão afetado: 50%.
- Ambos os pais afetados: 50% de risco de acondroplasia heterozigota, 25% risco de acondroplasia homozigota e 25% de chance de filho não afetado.
- Mutação De novo (nova): risco de recorrência baixo.
Distrofia torácica asfixiante
Prevalência:
- 1 em 70.000 nascimentos.
Diagnóstico Ultrassonográfico:
- A expressão fenotípica varia. Características típicas são tórax curto e estreito com costelas curtas e leve a moderado encurtamento rizomélico (fêmur e úmero) de membros que se tornam aparentes >22 semanas.
- A condição também é conhecida como síndrome de Jeune.
Anormalidades associadas:
- Anomalias renais.
Investigação:
- Ultrassonografia detalhada.
- Mutações em >10 genes foram identificadas, mas 50% dos casos ocorrem devido à mutação no gene DYNC2H1.
Seguimento:
- Rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Mortalidade neonatal: 60% por hipoplasia pulmonar.
- Os neonatos sobreviventes têm inteligência normal, mas raramente vivem além dos anos da adolescência por conta de problemas renais e cardiovasculares.
Recorrência:
- Herança autossômica recessiva: 25%.
Atelosteogênese
Prevalência:
- 1 em 500.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Encurtamento grave dos membros, tórax estreito, pé torto, luxação dos quadris, joelhos e cotovelos.
- Associada a testa proeminente, hipertelorismo, micrognatia e fenda palatina e “polegar de caroneiro”.
Investigação:
- Ultrassonografia detalhada.
- Atelosteogênese é causada por mutações nos genes FLNB e SLC26A2.
- Se a mutação na família é conhecida, diagnóstico pré-natal é melhor realizado através da biópsia vilo coriônica e análise de DNA.
Seguimento:
- Se a gravidez continuar, rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Letal intra-útero ou logo após o parto por falência respiratória.
Recorrência:
- Na maioria dos casos (mutações novas): não há risco aumentado.
- Nos casos raros de herança autossômica recessiva: 25%.
Displasia campomélica
Prevalência:
- 1 em 200.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Encurtamento e arqueamento dos ossos longos das pernas (angulação bilateral grave do fêmur), tórax estreito, escápulas hipoplásicas, crânio grande com face desproporcionalmente pequena, micrognatia.
Anormalidades associadas:
- Edema nucal, pé torto, hidrocefalia (devido à oclusão atlanto-occipital). Polidramnia é comum.
Investigação:
- Ultrassonografia detalhada.
- Há alteração do sexo em cerca de 70% dos fetos masculinos e teste invasivo é necessário para determinação do sexo genético.
- Displasia Campomélica é causada por mutações no ou próximas ao gene SOX9.
Seguimento:
- Se a gravidez continuar, rotineiro.
Parto:
- Assistência obstétrica de rotina e parto em centro terciário.
Prognóstico:
- Mortalidade no primeiro ano de vida: 95% devido à laringotraqueomalácia grave.
Recorrência:
- Na maioria dos casos (mutações novas): não há risco aumentado.
- Nos casos raros de herança autossômica recessiva: 25%.
Craniossinostose
Prevalência:
- 1 em 2.000 nascimentos.
- Associada à idade paterna avançada.
Diagnóstico Ultrassonográfico:
- Fusão prematura das suturas cranianas resultando em formas anormais do crânio.
Anormalidades associadas:
- Em 90% dos caso, craniossinostose é achado isolado.
- Em 10% dos casos, há associação com qualquer uma de 150 síndromes incluindo, síndrome de Crouzon, síndrome de Muenke, síndrome de Saethre-Chotzen, síndromes de Apert e síndrome de Pfeiffer.
Investigação:
- Ultrassonografia detalhada. Deve-se dar atenção especial para as mãos, área central da face, coração e sistema nervoso central.
- RMN oferece informações úteis.
- História familiar detalha e consulta com geneticista: se história familiar negativa para craniossinostose isolada, é recomendado cariótipo para excluir anormalidades cromossômicas.
- Craniossinostose é causada por mutações nos genes FGFR-2, FGFR-3, TWIST e EFNB-1.
Seguimento:
- Rotineiro.
Parto:
- Assistência obstétrica de rotina e parto em centro terciário.
Prognóstico:
- Recém-nascidos podem apresentar dificuldades para respirar, se alimentar e de visão.
- Pressão intracraniana aumentada, que é vista em um terço dos neonatos, pode afetar a inteligência e neurodesenvolvimento. Momento para reparo cirúrgico deve ser bem planejado.
Recorrência:
- Mutações novas: não há risco aumentado.
- Formas sindrômicas com história familiar positiva: 25% e 50% de risco dependendo da síndrome associada.
Síndrome Crouzon
Prevalência:
- 1 em 25.000 nascimentos.
Diagnóstico ultrassonográfico:
- Craniossinostose variável ( mais comumente bicoronal), hipoplasia da face média com nariz pontudo, prognatismo mandibular e exorbitismo (protrusão de globos oculares como resultado de órbitas rasas).
- Mãos e pés normais.
Investigação:
- Herança autossômica dominante da mutação do gene FGFR2 . O diagnóstico pode ser feito por teste invasivo.
Prognóstico:
- A maioria dos indivíduos afetados têm inteligência normal.
- Olhos protrusos e problemas de visão.
Síndrome Muenke
Prevalência:
- 1 em 30.000 nascimentos.
Diagnóstico ultrassonográfico:
- Cranissinostose coronal.
- Anormalidades leves de mãos e pés, incluindo fusão carpal e tarsal, braquidactilia, falange média em dedal e epífises cônicas.
Investigação:
- Herança autossômica dominante da mutação do gene FGFR3 . O diagnóstico pode ser feito por teste invasivo.
Prognóstico:
- A maioria dos indivíduos afetados é de inteligência normal, mas alguns apresentam retardo no desenvolvimento e dificuldades de aprendizado.
Síndrome Saethre-Chotzen
Prevalência:
- 1 em 40.000 nascimentos.
diagnóstico ultrassonográfico:
- Craniossinostose uni ou bicoronal, assimetria facial, desvio de septo nasal, implantação baixa da linha frontal capilar e orelhas pequenas e de baixa implantação, rodadas posteriormente.
- Braquidactilia e sindactilia cutânea parcial de dedos dos pés.
Investigação:
- Herança autossômica dominante da mutação do gene TWIST1 . O diagnóstico pode ser feito por teste invasivo.
Prognóstico:
- A maioria dos indivíduos afetados é de inteligência normal, mas alguns apresentam retardo no desenvolvimento e dificuldades de aprendizado.
Síndrome de Apert
Prevalência:
- 1 em 70.000 nascimentos.
Diagnóstico ultrassonográfico:
- Craniossinostose bicoronal resultando em braquicefalia, deformidade do crânio em torre, hipertelorismo, bossa frontal.
- Braquissindactilia de mãos e pés.
- Outras anormalidades: defeitos de septo ventricular, fusão de vértebras cervicais, agenesia de corpo caloso, hidronefrose e criptorquidia.
Investigação:
- Herança autossômica dominante da mutação do gene FGFR2 . O diagnóstico pode ser feito por teste invasivo.
Prognóstico:
- 50% de chance de retardo mental.
- Necessidade de cirurgia para sinostose e sindactilia.
Síndrome de Pfeiffer
Prevalência:
- 1 em 70.000 nascimentos.
Diagnóstico ultrassonográfico :
- Craniossinostose de múltiplas suturas ( crânio trevo) e hipoplasia de face média com hipertelorismo associado e exorbitismo.
- Polegares largos e desviados radialmente, hálux largo.
Investigação:
- Herança autossômica dominante da mutação do gene FGFR1 e FGFR2. O diagnóstico pode ser feito por teste invasivo.
Prognóstico:
- Tipo I: prognóstico favorável, inteligência normal.
- Tipo II: prognóstico neurológico ruim.
Displasia diastrófica
Prevalência:
- 1 em 500.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Expressão fenotípica variável. Características típicas incluem encurtamento grave e arqueamento de todos os ossos longos, contratura em flexão de múltiplas articulações, escoliose, pé torto e polegares em abdução (“polegar do caroneiro”).
Anormalidades associadas:
- Micrognatia, fenda palatina e orelhas espessadas e deformadas ( “em couve-flor”).
Investigação:
- Ultrassonografia detalhada.
- Displasia diastrófica é uma das diversas desordens esqueléticas causadas por mutações no gene SLC26A2.
Seguimento:
- Rotineiro.
Parto:
- Assistência obstétrica de rotina e parto em centro terciário.
Prognóstico:
- A doença não é letal e o neurodesenvolvimento é normal.
- Problemas médicos a longo prazo incluem estatura baixa, escoliose progressiva e deformidades nas articulações dificultando a marcha.
Recorrência:
- Herança autossômica recessiva: 25%.
Síndrome Ellis-Van Creveld
Prevalência:
- 1 em 100.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Encurtamento acromélico e mesomélico dos membros, polidactilia pós-axial, tórax estreito, displasia ectodérmica e defeitos cardíacos.
Anormalidades associadas:
- Defeitos cardíacos estão presentes em >50% dos casos: átrio único, defeitos de septo atrial grande, atresia aórtica, aorta ascendente hipoplásica e hipoplasia de ventrículo esquerdo.
Investigação:
- Ultrassonografia detalhada.
- A síndrome de Ellis-Van Creveld é causada por mutações no gene EVC ou EVC2.
Seguimento:
- Rotineiro.
Parto:
- Assistência obstétrica de rotina e parto em centro terciário.
Prognóstico:
- Mortalidade neonatal: 50% devido a hipoplasia pulmonar e problemas cardíacos.
- O prognóstico final é relacionado à presença ou ausência de anormalidades cardíacas. Inteligência é normal.
Recorrência:
- Herança autossômica recessiva: 25%.
Hipofosfatasia
Prevalência:
- Hipofosfatasia é desordem herdada que afeta o desenvolvimento ósseo e dentário. Pode aparecer a qualquer momento, desde antes do nascimento até a vida adulta.
- Tipo Pré-Natal: 1 em 100.000 nascimentos.
- Tipo Pós-Natal: 1 em 10.000 nascimentos.
Diagnóstico Ultrassonográfico:
- No tipo pré-natal há micromelia grave, hipoplasia torácica severa, hipomineralização difusa ( ecogenicidade diminuída) acometendo todos os ossos exceto as clavículas.
Investigação:
- Ultrassonografia detalhada.
- Hipofosfatasia é causada por mutações no gene ALPL que levam à produção de versão anormal da fosfatase alcalina que não pode participar de forma efetiva do processo de mineralização. O grau de disfunção/gravidade varia desde casos não aparentes com diagnóstico através de características bioquímicas até casos letais detectados no feto.
- Se a mutação na família é conhecida, diagnóstico pré-natal é melhor realizado através da biópsia vilo coriônica e análise de DNA.
Seguimento:
- Se a gravidez continuar, rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Tipo Pré-Natal: letal devido à grave hipoplasia pulmonar.
- Tipo Pós-Natal: pode aparecer na infância ou idade adulta. Crianças podem apresentar baixa estatura, pernas arqueadas e geno valgo, articulação do punho e tornozelo aumentada e forma anormal do crânio. Nos adultos, há perda prematura de dentes e fraturas recorrentes nos pés e fêmur causando dor crônica.
Recorrência:
- Tipo pré-natal é autossômico recessivo: 25%
- Tipo pós-natal é ou autossômico recessivo ou dominante: 25-50%.
Síndrome Jarcho-Levin
Prevalência:
- 1 em 200.000 nascimentos.
- Também conhecida como disostose espôndilo-torácica e nanismo de tronco curto.
Diagnóstico Ultrassonográfico:
- Fusão de vários corpos vertebrais e desalinhamento com as costelas que estão também fundidas na porção próxima à coluna. O tronco é curto, mas braços e pernas têm comprimento normal.
Investigação:
- Ultrassonografia detalhada.
- Síndrome de Jarcho-Levin é causada por mutações no gene MESP2.
Seguimento:
- Rotineiro.
Parto:
- Assistência obstétrica de rotina e parto em centro terciário pois o neonato pode precisar de ressuscitação devido à hipoplasia pulmonar.
Prognóstico:
- Cerca de 40% morre nos primeiros meses devido à hipoplasia pulmonar e hipertensão. Após o segundo ano de vida a condição melhora. Neurodesenvolvimento é normal.
Recorrência:
- Herança autossômica recessiva: 25%.
Osteogênese imperfeita
Prevalência:
- 1 em 15.000 nascimentos.
- As mais comuns são tipo I e IV.
Diagnóstico Ultrassonográfico:
- Espectro de defeitos caracterizado por ossos frágeis.
- Há pelo menos oito tipos de osteogênese imperfeita do tipo I ao VIII com características que se sobrepõe. Tipo I é a forma mais leve e II a mais grave; os sinais e sintomas dos outros tipos estão entre esses dois extremos.
- Os casos mais comuns de diagnóstico pré-natal são tipos II e III:
- Tipo I: ossos frágeis, esclera azul e surdez progressiva. Ultrassonografia no segundo e terceiro trimestres pode demonstrar fratura de ossos longos.
- Tipo II: membros e costelas curtas com múltiplas fraturas, hipomineralização do crânio.
- Tipo III: múltiplas fraturas, usualmente presentes ao nascimento, resultando em escoliose e estatura muito baixa.
Investigação:
- Ultrassonografia detalhada.
- História familiar detalhada e consulta com geneticista.
- Osteogênese imperfeita é causada por mutações nos genes COL1A1, COL1A2, CRTAP e P3H1.
- Diagnóstico pré-natal dos tipos II, III e IV pode ser feitos por teste invasivo.
Seguimento:
- Rotineiro.
Parto:
- Assistência obstétrica de rotina e parto em centro terciário
Prognóstico:
- Tipo I: expectativa de vida normal.
- Tipo II: letal.
- Tipo III deficiência motora ( cifoscoliose, fraturas), perda auditiva na vida adulta.
Recorrência:
- A maioria dos casos de osteogênese imperfeita tem padrão de herança autossômico dominante, mas a maioria das crianças com formas mais graves ( tipo II e III) tem doença causada por mutações novas.
Síndrome de costela curta e polidactilia
Prevalência:
- 1 em 500.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Micromelia, costelas curtas e tórax hipoplásico, polidactilia ( geralmente pré-axial).
- Há 4 tipos da síndrome:
- Tipo I ( Saldino-Noonan): metáfises estreitas.
- Tipo II (Majewski): fenda facial e tíbia desproporcionalmente curta.
- Tipo III (Naymoff): metáfises largas com esporões.
- Tipo IV (Beermer-Langer): fenda labial mediana e genitália ambígua em alguns indivíduos 46,XY.
Anormalidades associadas:
- Defeitos cardíacos, rins policístico, malformações cerebrais e atresia intestinal.
Investigação:
- Ultrassonografia detalhada.
Seguimento:
- Se a gravidez continuar, rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Condição letal devido à grave hipoplasia pulmonar.
Recorrência:
- Herança autossômica recessiva: 25%.
Displasia tanatofórica
Prevalência:
- 1 em 10.000 nascimentos.
- Displasia esquelética letal mais comum.
Diagnóstico Ultrassonográfico:
- Encurtamento grave dos membros, tórax estreito, comprimento do tronco normal e cabeça grande com testa proeminente.
- Há 2 tipos de displasia tanatofórica:
- Tipo I (mais comum): esporádica; fêmur curvado ( “em telefone”).
- Tipo II (raro): esporádica, o fêmur é reto porém o crânio é em forma de trevo.
Investigação:
- Ultrassonografia detalhada.
- Displasia tanatofórica é causada por mutação no gene FGFR3. O diagnóstico pode ser feito por teste invasivo ou análise de cfDNA no sangue materno.
Seguimento:
- Se a gravidez continuar, rotineiro.
Parto:
- Assistência obstétrica e parto de rotina.
- As gestações são em geral, complicadas por polidramnia, prematuridade, apresentações anômalas e desproporção céfalo-pélvica . A presença de crânio em trevo e hidrocefalia podem levar a necessidade de cefalocentese e assistência ao parto.
Prognóstico:
- A condição é letal devido à hipoplasia pulmonar grave.
Recorrência:
- Não há risco aumentado.
Hidropsia fetal
Tópicos nessa seção
- Panorama
Panorama
Prevalência:
- 1 em 2.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Acúmulo anormal de líquido seroso em pelo menos dois dos seguintes: pele (edema) e cavidades corpóreas (derrames pericárdico, pleural ou ascite).
- Placentomegalia ( espessura placentária > 6cm) é vista com frequência.
Etiologia:
- Achado não específico em grande variedade de desordens maternas e fetais, incluindo anomalias hematológicas, cromossômicas, cardiovasculares, renais, pulmonares, gastrointestinais, hepáticas e metabólicas, infecções congênitas, neoplasmas, malformações da placenta ou cordão umbilical e síndromes genéticas.
- Classicamente dividida em:
- Imune: 10% dos casos; devido a anticorpos hemolíticos maternos.
- Não Imune: 90%; devido a outras etiologias.
Investigação:
- Em várias situações, a causa subjacente pode ser determinada por rastreio infeccioso e de anticorpos maternos, ultrassonografia incluindo ecocardiograma, Doppler e amostra de sangue fetal e líquido amniótico para cariótipo e array.
- Frequentemente, a anomalia continua inexplicada mesmo após necropsia realizada por especialista.
Terapia Fetal:
- Anemia fetal: transfusões intrauterinas.
- Derrame pleural ou grande cisto pulmonar: inserção de shunt tóraco-amniótico.
- Taquiarritmia fetal: administração transplacentária ou diretamente ao feto de drogas antiarrítmicas.
- Teratomas, corioangiomas e sequestro pulmonar: coagulação do vaso alimentador através de laser guiado por ultrassom.
- Feto receptor na síndrome de transfusão feto-fetal: coagulação dos vasos placentários comunicantes por laser endoscópico.
Seguimento:
- Ultrassom a cada 2-3 semanas para monitorar evolução da hidropsia.
- Há risco de morbidade materna devido à “síndrome espelho” ( combinação de hidropsia fetal com sobrecarga hídrica generalizada e estado pré-eclâmptico na mãe).
Parto:
- Momento e via vão depender da causa da hidropsia.
Prognóstico:
- Depende da causa da hidropsia.
- Hidropsia progressiva inexplicada é, em geral, letal antes ou logo após o nascimento.
Recorrência:
- Defeitos fetais, infecção: não há risco aumentado.
- Parte de trissomia: 1%.
- Isoimunização: alta.
- Desordens metabólicas: 25%.
Defeitos cromossômicos
Tópicos nessa seção
- Características
- Manejo
Defeitos cromossômicos
Características
Os mais comuns defeitos cromossômicos são as trissomia 21,18 e 13, defeitos cromossômicos sexuais (45,X, 47,XXY, 47XYY) e triploidia.
No primeiro trimestre, uma característica de muitos defeitos cromossômicos é espessura aumentada da translucência nucal. Com o avançar da gestação, cada defeitos cromossômico tem seu próprio padrão sindrômico de anormalidades.
- Trissomia 21:
Hipoplasia nasal, prega pré-nasal e nucal aumentadas, defeitos cardíacos, focos ecogênicos intracardíacos, atresia duodenal e intestino hiperecogênico, hidronefrose leve, encurtamento do fêmur, afastamento do hálux (“sandal gap”) e Clinodactilia ou hipoplasia da falange média do quinto dedo.
- Trissomia 18:
Crânio em forma de morango, cistos de plexo coroide, ausência de corpo caloso, cisterna magna aumentada, fenda facial, micrognatia, edema nucal, problemas cardíacos, hérnia diafragmática, atresia esofagiana, onfalocele, artéria umbilical única, anormalidades renais, intestino hiperecogênico, mielomeningocele, crescimento restrito e encurtamento de membros, aplasia radial, dedos sobrepostos e pé torto ou pés em cadeira de balanço.
- Trissomia 13:
Holoprosencefalia, microcefalia, anormalidades faciais e cardíacas, rins aumentados e hiperecogênicos, onfalocele e polidactilia pós-axial.
- Triploidia:
Quando há dupla contribuição paterna (diândrica) há placenta molar e a gravidez raramente evolui após 20 semanas. Quando há contribuição materna (digínica) a placenta é fina porém de consistência normal e a gravidez pode chegar até o terceiro trimestre; o feto apresenta grave crescimento restrito assim étrico, ventriculomegalia leve, micrognatia, anormalidades cardíacas, mielomeningocele, sindactilia e hálux em abdução ( “dedão do caroneiro”).
- Síndrome de Turner:
Grandes higromas císticos, edema generalizado, derrame pleural e ascite leves, anormalidades cromossômicas e rim em ferradura.
- 47,XXX, 47XXY ou 47,XYY:
Essas não são associadas à elevada prevalência de defeitos detectáveis a ultrassonografia.
Manejo
Quando uma anormalidade sugestiva de defeitos cromossômico é detectada, uma avaliação completa deve ser feita à procura de outras características do defeito cromossômico que sabidamente se associam a tal marcador.
- Anormalidades Múltiplas:
O risco de anomalias cromossômicas é muito aumentado e cariótipo fetal deve ser considerado.
- Anormalidades Isoladas:
A decisão de prosseguir ou não com teste invasivo depende se a anormalidade é maior ou menor.
- Se houver anormalidade maior, é aconselhável oferecer cariotipagem, mesmo que essas sejam aparentemente isoladas. Isso pois: primeiramente, se as anormalidades forem ou letais ou associadas a deficiência grave (p.ex. holoprosencefalia), cariotipagem fetal, constitui uma em uma série de investigações para determinar a possível causa e logo, o risco de recorrência; em segundo lugar, caso a anormalidade seja potencialmente corrigível cirurgicamente no período intrauterino ou pós-natal (p.ex. hérnia diafragmática), é importante excluir defeito cromossômico – especialmente porque, para várias dessas condições, o defeito usual é trissomia 18 ou 13.
- Se houver anormalidade menor, o risco pode ser estimado multiplicando-se o risco a priori (baseado nos resultados do rastreio anterior) pelo risco relativo de cada marcador ou anormalidade específico. O risco relativo para trissomia 21 é cerca de 1 (logo, o risco a priori não é aumentado em caso de cisto de plexo coróide, foco ecogênico intracardíaco, hidronefrose leve e fêmur curto) e cerca de 10 ( logo há aumento de 10 vezes no risco a priori para edema nucal ou pré-nasal e osso nasal hipoplásico).
Tumores
Tópicos nessa seção
- Linfangioma
- Neuroblastoma
Linfangioma
Prevalência:
- 1 em 6.000 nascimentos.
Diagnóstico Ultrassonográfico:
- Massa irregular, multisseptada, multicística, que usualmente é localizada no pescoço(75%), região axilar (20%), parede torácica, abdominal ou extremidades (5%); em < 1% dos casos, o tumor se encontra no mesentério ou retroperitônio.
Anormalidades associadas:
- A incidência de anomalias cromossômicas ou síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada.
- Em casos de linfangioma cervical ou torácico, há risco aumentado de compressão venosa central levando à hidropsia e compressão esofágica com polidramnia.
Seguimento:
- Ultrassonografia a cada 2-3 semanas para monitorar tamanho do tumor e volume de líquido amniótico. Amniodrenagem pode ser necessária em caso de polidramnia e encurtamento do colo.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: cesariana e procedimento EXIT caso haja polidramnia e hiperextensão do pescoço por grande tumor cervical.
Prognóstico:
- Prognóstico é favorável, a menos que em associação à falência cardíaca, hidropsia ou compressão de via aérea.
- Algumas lesões mesenquimais podem regredir espontaneamente de forma parcial após o nascimento. O tratamento de escolha é excisão cirúrgica.
Recorrência:
- Não há risco aumentado.
Neuroblastoma
Prevalência:
- 1 em 20.000 nascimentos.
- Surge de tecido neural indiferenciado da medula adrenal (90%) ou gânglios simpáticos no abdome, tórax, pelve ou cabeça e pescoço (10%).
Diagnóstico Ultrassonográfico:
- Massa cística, sólida ou complexa na região da glândula adrenal, usualmente no terceiro trimestre. Ocasionalmente, calcificações estão presentes.
- Tumores advindos de gânglios simpáticos podem aparecer no abdome, tórax e pescoço.
- O tumor pode metastatizar in utero para placenta, cordão umbilical ou fígado.
- Pode haver polidramnia ou hidropsia fetal associada.
Anormalidades associadas:
- A incidência de anomalias cromossômicas ou síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada.
- RMN para detectar possível extensão intra-espinhal do tumor.
Seguimento:
- Ultrassom a cada 4 semanas para monitorar tamanho do tumor e desenvolvimento de hepatomegalia, placentomegalia, falência cardíaca e polidramnia.
Parto:
- Local: hospital com UTI neonatal e cirurgia pediátrica.
- Momento: 38 semanas.
- Via: indução de parto normal.
Prognóstico:
- Sobrevivência é > 90% se o diagnóstico for feito in utero ou no primeiro ano de vida, mas < 20% para os diagnosticados após o primeiro ano. Tratamento requer excisão cirúrgica do tumor e quimioterapia.
Recorrência:
- Não há risco aumentado.
Placenta, cordão umbilical
Tópicos nessa seção
- Corioangioma
- Acretismo placentário
- Artéria umbilical única
- Cisto de cordão umbilical
- Nó de cordão umbilical
- Vasa previa
Corioangioma
Prevalência:
- 1 em 5.000 gestações.
Diagnóstico Ultrassonográfico:
- Massa bem circunscrita hipo ou hiperecóica que, em geral, está localizada abaixo da placa coriônica próximo a inserção do cordão umbilical e usualmente se projeta para cavidade amniótica.
- Color Doppler demonstra grandes canais vasculares ao redor e dentro do tumor.
Anormalidades associadas:
- Grandes tumores podem resultar em anemia fetal e trombocitopenia ( devido a sequestro de células vermelhas e plaquetas pelo tumor), insuficiência cardíaca fetal, hidropsia e placentomegalia (devido à circulação hiperdinâmica como resultado de shunt arteriovenoso), polidramnia (por transudação direta para líquido amniótico e poliúria fetal secundária à circulação hiperdinâmica) e síndrome espelho materna ( sobrecarga fluida generalizada e pré-eclâmpsia).
Investigação:
- Ultrassonografia detalhada, incluindo ecocardiograma para avaliação da função cardíaca e medida do pico de velocidade sistólica da artéria cerebral média (PVS ACM) para diagnóstico de anemia fetal.
Seguimento:
- Ultrassom a cada 2-3 semanas para monitorar crescimento do tumor, função cardíaca, PVS ACM e volume de líquido amniótico.
- Coagulação a laser dos vasos do tumor guiada por ultrassom, transfusões fetais e Amniodrenagem podem ser necessárias.
Parto:
- Local: hospital com UTI neonatal.
- Momento: 38 semanas. Mais precocemente se houver evidência de restrição de crescimento, hipóxia fetal ou hidropsia.
- Via: indução de parto vaginal, a não ser que o feto esteja hidrópico ou hipóxico.
Prognóstico:
- Corioangiomas sintomáticos apresentam risco de morte perinatal. O neonato pode ter grave anemia hemolítica microangiopática e trombocitopenia.
Recorrência:
- Não há risco aumentado.
Acretismo placentário
Prevalência:
1 em 400 gestações. • Placenta prévia com história de parto cesáreo (PC) anterior: 3% para 1 PC, 10% para 2 PC, > 50% para ? 3 PC. • Acretismo placentário inclui placenta acreta ( vilosidades coriônicas aderidas ao miométrio), increta ( vilosidades invadem o miométrio) e percreta ( vilosidades ultrapassam o miométrio).
Diagnóstico Ultrassonográfico:
- Múltiplas lacunas vasculares ( espaços) na placenta ( aparência em “queijo suíço”) com fluxo turbilhonar ( pico de velocidade sistólica > 15cm/s).
- Espessura miometral retroplacentária de < 1mm.
- Perda da hipoecogenicidade normal da zona retroplacentária.
- Vasos sanguíneos e tecido placentário cruzando a margem útero-placentária, interface miométrio-vesical ou cruzando a serosa uterina. Massas exofíticas invadindo a bexiga urinária.
- Vascularização irregular envolvendo toda a junção serosa uterina/bexiga, visualizada em power Doppler 3D.
Investigação:
- RMN é recomendada caso os achados ultrassonográficos sejam inconclusivos.
- Vigilância pré-natal fetal deve ser rotineira.
Seguimento:
- Rotineiro.
Parto:
- Local: hospital com experiência no manejo dessa condição e banco de sangue que possa facilitar transfusão de grandes quantidades de hemoderivados. Há alta chance de histerectomia e hemorragia grave.
- Momento: 36-37 semanas.
- Via: Cesariana.
Prognóstico:
- Mortalidade materna 5-10%, morbidade 75%.
- Diagnóstico precoce reduz a mortalidade e morbidade em 50%.
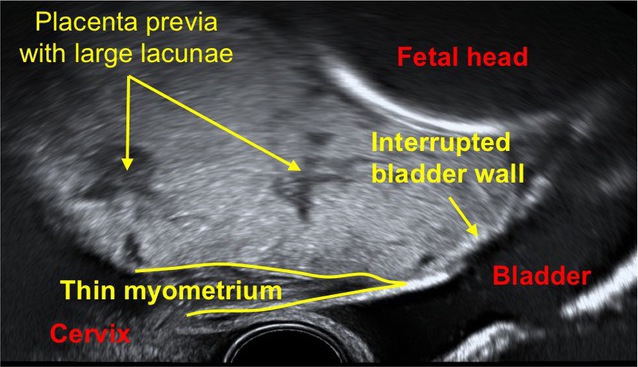
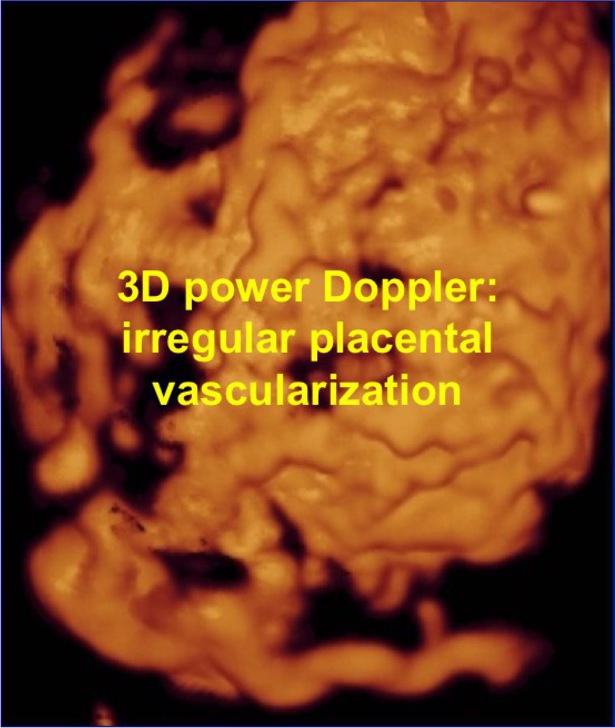
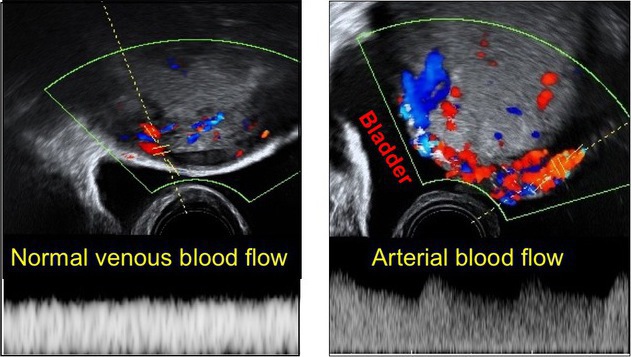
Artéria umbilical única
Prevalência:
- 1 em 100 gestações.
Diagnóstico Ultrassonográfico:
- Visualização de apenas 1 artéria umbilical ao redor da bexiga fetal.
Anormalidades associadas:
- Anomalias cromossômicas, principalmente trissomia 18, 13 e triploidia, são vistas em 5% dos casos.
- Restrição de crescimento fetal (< percentil 5) ocorre em 10% dos casos. Morte fetal intrauterina, em geral associada a restrição de crescimento, é 2 vezes mais comum do que na população geral.
- Anormalidades afetando sistemas cardiovascular, esquelético, gastrointestinal, geniturinário e nervoso central, são vistas em 20% dos casos.
Investigação:
- Ultrassonografia detalhada.
- Cariótipo fetal nos casos não isolados.
Seguimento:
- Ultrassom seriado com 28,32 e 36 semanas para avaliar crescimento e bem-estar fetal.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Em casos isolados, o prognóstico é normal.
Recorrência:
- Isolada: não há risco aumentado.
- Parte de trissomias: 1%.
Cisto de cordão umbilical
Prevalência:
- 1 em 100 gestações.
Diagnóstico Ultrassonográfico:
- Cistos verdadeiros são derivados de remanescentes embriológicos ou do alantoide ou do ducto onfalomesentérico e são tipicamente localizados próximos a inserção fetal do cordão.
- Pseudocistos são mais comuns que cistos verdadeiros e se localizam em qualquer lugar ao longo do cordão. Não apresentam revestimento endotelial e representam edema localizado e liquefação da geleia de Wharton.
Anormalidades associadas:
- Cistos únicos são geralmente transitórios com nenhum efeito adverso.
- Múltiplos cistos são associados a maior risco de abortamento, trissomia 18 e 13, onfalocele, associação VACTREL e restrição de crescimento fetal.
Investigação:
- Ultrassonografia detalhada.
- Cariótipo fetal em casos não isolados.
Prognóstico:
- Casos isolados: prognóstico normal.
Recorrência:
- Casos isolados: não há risco aumentado.
- Parte d trissomias: 1%
Nó de cordão umbilical
Prevalência:
- 1 em 100 gestações.
Diagnóstico Ultrassonográfico:
- Visualização de corte transversal do cordão rodeado por alça de cordão: “sinal do laço de corda”.
- Fatores predisponentes incluem: cordão longo, feto pequeno, polidramnia, gêmeos monoamnióticos.
Investigação:
- Ultrassonografia detalhada.
- Avaliação rotineira do cordão à procura de nós pode não ser justificável por ser demorada e de sensibilidade e benefício duvidosos.
Seguimento:
- A detecção de nó de cordão umbilical não indica parto a não ser que associado a evidência de comprometimento do bem-estar fetal como padrão anormal de frequência cardíaca.
- Deve-se considerar monitoramento fetal através de Doppler e cardiotocografia.
Prognóstico:
- Risco de morte intrauterina é aumentado em 5 vezes.
Recorrência:
- Não há risco aumentado.
Vasa previa
Prevalência:
- 1 em 3.000 gestações.
Diagnóstico Ultrassonográfico:
- Vasos umbilicais, não contidos pelo cordão umbilical ou tecido placentário, atravessam as membranas no segmento inferior logo acima do colo.
- Uso de ultrassonografia transvaginal e color Doppler são necessários para o diagnóstico.
Anormalidades associadas:
- Vasa previa em geral, ocorre em associação à inserção velamentosa do cordão, placenta bilobata ou succenturiada, em que os vasos passam ao longo das membranas para juntar os lobos.
- Fatores de risco são gestações múltiplas, concepção por FIV (1 em 300) e placenta baixa no segundo trimestre.
Investigação:
- Ultrassonografia detalhada, incluindo exame transvaginal com color Doppler.
Seguimento:
- Ultrassom seriado a cada 2 semanas após 26 semanas para monitorar comprimento do colo. Se houver encurtamento cervical (< 25 mm), deve-se considerar hospitalização.
Parto:
- Parto cesáreo com 34-36 semanas dependendo do comprimento cervical.
Prognóstico:
- Morte fetal: >60% caso não seja diagnosticada no pré-natal e <3% se for.
Recorrência:
- Não há risco aumentado.
Líquido amniótico
Tópicos nessa seção
- Oligodramnia
- Polidramnia
Oligodramnia
Prevalência:
- 1 em 100 gestações < 24 semanas.
Diagnóstico Ultrassonográfico:
- Medida do maior bolsão vertical de líquido amniótico livre de partes fetais é < 2cm ou índice de líquido amniótico ( soma dos bolsões verticais dos 4 quadrantes) é < 5cm.
Anormalidades associadas:
- Existem basicamente 3 grandes causas para oligodramnia com < 24 semanas:
- Anormalidades do Trato Urinário: agenesia renal bilateral, rins multicísticos ou policísticos e obstrução uretral.
- Rotura prematura de membranas pré-termo: crescimento fetal, anatomia e Doppler normais com história materna de perda vaginal de líquido claro ou “tingido”de sangue.
- Insuficiência útero-placentária: Restrição de crescimento fetal com evidencia ao Doppler de resistência ao fluxo nas artérias uterinas e/ou umbilicais e redistribuição da circulação fetal.
Investigação:
- Ultrassonografia detalhada.
- Em casos de oligodramnia inexplicada, amnioinfusão pode ser útil por permitir exame detalhado do feto e em alguns casos demonstrar que a causa é rotura de membranas.
- Teste invasivo para cariótipo fetal deve ser realizado caso haja anormalidades fetais relevantes.
Seguimento:
- Ultrassom a cada 1-3 semanas para monitorar condição fetal e avaliar líquido. Em caso de rotura de membranas, avaliação de crescimento pulmonar pode ser útil na predição de hipoplasia.
- Amnioinfusão terapêutica não é útil.
- Em casos de insuficiência útero-placentária, avaliação do crescimento fetal, Doppler da artéria umbilical, ducto venoso e artéria cerebral média ajudam a decidir o o melhor momento para o parto.
Parto:
- Anormalidades de trato urinário: assistência obstétrica e parto de rotina.
- Rotura de membranas: conduta expectante e parto vaginal se apresentação cefálica.
- Insuficiência útero-placentária: cesariana ou parto vaginal dependendo da idade gestacional, tamanho do feto, grau de comprometimento defino pelo Doppler ou cardiotocografia.
Prognóstico:
- Depende da idade gestacional no momento do diagnóstico, causa e idade gestacional no parto. Em oligodramnia < 24 semanas, o prognóstico, em geral, é ruim.
- Agenesia renal bilateral, rins multicísticos ou policístico são anomalias letais, usualmente no período neonatal por hipoplasia pulmonar.
- Rotura prematura de membranas ? 20 semanas é associada a prognóstico reservado; cerca de 40% abortam nos primeiros 5 dias devido à corioamnionite e, das 60% restantes, mais de 50% dos neonatos morre devido à hipoplasia pulmonar.
- Insuficiência útero-placentária levando a oligodramnia ? 24 semanas é muito grave e o desfecho mais provável é a morte fetal intrauterina.
Recorrência:
- Anormalidades renais: agenesia ou multicística: 1-3%; policística infantil 25%.
- Rotura prematura de membranas: 10-25% mas pode ser reduzida com cerclagem e progesterona.
- Insuficiência placentária: 10%, mas pode ser reduzida com uso de aspirina (150mg/dia) a partir de 12 semanas.
Polidramnia
Prevalência:
- 1 em 100 gestações.
Diagnóstico Ultrassonográfico:
- A medida do maior bolsão vertical de líquido amniótico livre de parte fetais é usada para classificar polidramnia em leve (8-11cm), moderada (12-15cm) e grave (?16cm).
- Em cerca de 80% dos casos a polidramnia é leve, em 15% moderada e 5% grave.
- A maioria dos casos de polidramnia leve são idiopáticos, mas a maioria das moderadas e graves ocorrem por desordens maternas ou fetais.
- Na maioria dos casos, polidramnia ocorre no final do segundo trimestre ou no terceiro trimestre. Polidramnia aguda com 16-22 semanas é vista principalmente associada à síndrome de transfusão feto-fetal.
Anormalidades associadas:
- Há, essencialmente, 2 causas para polidramnia:
- Diminuição da deglutição fetal: devido a anormalidade cerebrais (p.ex. anencefalia, malformação de Dandy-Walker), tumores faciais, obstrução gastrointestinal (p.ex. atresia esofagiana ou duodenal, obstrução de delgado), desordens pulmonares obstrutivas ( p.ex. derrame pleural, hérnia diafragmática, MPC, SCOVAA), caixa torácica estreita (devido a displasias esqueléticas) e sequencia deformativa de acinesia fetal (devido a déficit neuromuscular de deglutição fetal).
- Aumento da micção fetal: diabetes mellitus e uremia maternos ( aumento de glicose e ureia causam diurese osmótica), circulação fetal hiperdinâmica devido à anemia (p.ex. aloimunização ou infecção congênita), tumores fetais ou placentários (p.ex teratoma sacrococcígeo, corioangioma placentário), ou síndrome de transfusão feto-fetal.
Investigação:
- Ultrassonografia detalhada.
- Teste invasivo para cariótipo fetal e array caso haja anormalidades ou restrição de crescimento. Teste de DNA para mutação miotônica distrófica caso haja posicionamento anormal de extremidades.
- Teste de tolerância a glicose caso associada à macrossomia.
- TORCH se houver sinais fetais de infecção.
Seguimento:
- Ultrassom a cada 1-3 semanas para monitorar condição fetal, volume de líquido e comprimento cervical.
Terapia Pré-Natal:
- Diabetes mellitus materno: controle glicêmico.
- Hidropsia devido à arritmias: medicação antiarrítmica.
- Hidropsia por anemia fetal: transfusão intrauterina.
- Derrame pleural ou cisto pulmonar: shunt tóracoamniótico.
- Síndrome de transfusão feto-fetal: oclusão a laser de anastomoses placentárias.
- Tumores fetais ou placentários: oclusão a laser de vasos alimentadores.
- Defeitos que diminuem a deglutição fetal ou polidramnia idiopática grave: Amniodrenagem seriada caso haja encurtamento cervical. Entretanto o procedimento por si só pode precipitar parto prematuro. Uma alternativa e método eficaz de tratamento é administração materna de indometacina; porém essa droga pode causar contrição do ducto e monitoramento frequente com ecocardiograma seriado é necessário.
Parto:
- Assistência obstétrica e parto de rotina na maioria dos casos.
- Anormalidades fetais: indução do parto com 38 semanas em hospital com UTI neonatal e cirurgia pediátrica.
- Tumores fetais: considerar cesariana e procedimento EXIT.
- Polidramnia grave: indução e rotura de membranas controladas com 38 semanas para evitar o risco de prolapso de cordão.
Prognóstico:
- Depende da causa da polidramnia e da idade gestacional no parto.
Recorrência:
- Idiopática: não há risco aumentado.
- Associada a condições maternas e fetais: depende da causa.
Gestação múltipla
Tópicos nessa seção
- Protocolo para ultrassonografia
- Monocoriônicos: gemelaridade imperfeita
- Monocoriônicos: morte de um gemelar
- Monocoriônicos: monoamniótico
- Monocoriônicos: crescimento restrito seletivo
- Monocoriônicos: sequencia anemia-policitemia.
- Monocoriônicos: sequencia TRAP
- Monocoriônicos: síndrome de transfusão feto-fetal
- Anormalidades estruturais
- Gestação trigemelar
Gestação múltipla
Protocolos para ultrassonografia
Introdução:
- Parto gemelar contribui com 2-3% de todos os nascimento e a incidência está aumentando devido à concepção assistida e idade materna avançada.
- Mortalidade e morbidade perinatal são maiores nas gestações múltiplas do que nas únicas e por isso elas requerem suporte adicional.
- O risco de complicações é muito maior em gestações monocoriônicas (MC) do que em dicoriônicas (DC).
- Dois terços dos gêmeos são dizigóticos ( não idênticos) e um terço é monozigótico (idêntico). Um terço dos monozigóticos são dicoriônicos (DC) e dois terços são Monocoriônicos (MC). Logo, todos os gêmeos MC são monozigóticos, e 6 de 7 gêmeos DC são dizigóticos.
- Em gestações DC, a membrana inter-fetal é composta de camada central de tecido coriônico entre 2 camadas de amnio, enquanto nas gestações MC não há camada coriônica.
Determinação de corionicidade:
- A melhor maneira de determinar corionicidade pela ultrassonografia de 11-13 semanas é examinar a junção entre a membrana inter-fetal e a placenta. Em gestações DC, há projeção triangular de tecido placentário ( sinal ?) para base da membrana. Em gestações MC, não há projeção de tecido ( sinal T).
- Com o avançar da gravidez, há regressão do folheto coriônico e o sinal “lambda”se torna progressivamente mais difícil de identificar. Logo, com 20 semanas, apenas 85% das gestações DC demonstram sinal ?.
Datação da gravidez:
- Concepção espontânea: usar o comprimento cabeça-nádega do maior feto na avaliação de 11-13 semanas.
- Concepção por FIV: usar a idade embrionária da fertilização.
Medidas:
- Em cada ultrassom, avaliar o crescimento fetal ( circunferência cefálica e abdominal, comprimento do fêmur), líquido amniótico ( maior bolsão vertical), índice de pulsatilidade ao Doppler ( artéria umbilical, cerebral média e ducto venoso) e, em monocorônicos, pico de velocidade sistólica da ACM para detectar possível sequencia anemia-policitemia (TAPS).
- Com 20 semanas, medida do comprimento do colo. Se < 20mm, iniciar 400mg de progesterona pela manhã e 400mg a noite e reavaliar em 1 semana. Se houver encurtamento progressivo < 5mm, administrar corticoide profilático para amadurecimento pulmonar. Não há evidências de que repouso ou cerclagem sejam benéficos.
Gêmeos dicoriônicos:
- Ultrassom com 12 e 20 semanas e então a cada 4 semanas até o parto.
- Se houver discordância no peso fetal >15%, discordância de líquido amniótico ou Doppler anormal, reavaliar em 1 semana.
- Se não houver complicação, considerar parto com 37 semanas.
Gêmeos monocoriônicos diamnióticos:
- Ultrassom com 12 e 16 semanas e então a cada 2 semanas até o parto.
- Se houver discordância no peso fetal >15%, discordância de líquido amniótico ou Doppler anormal, reavaliar em 1 semana.
- Se não houver complicação, considerar parto com 36 semanas.
Gêmeos monocoriônicos monoamnióticos:
- Ultrassom com 12 e 16 semanas e então a cada 2 semanas até o parto.
- Se houver discordância no peso fetal >15% ou Doppler anormal, reavaliar em 1 semana.
- Se não houver complicação, parto cesáreo com 32 semanas.
Trigemelares tricoriônicos:
- 12 semanas: aconselhamento considerando opções de conduta expectante ou embriorredução.
- Ultrassom com 12,20,24,28 e 32 semanas.
- Se houver discordância no peso fetal >15%, discordância de líquido amniótico ou Doppler anormal, reavaliar em 1 semana.
- Se não houver complicação, parto cesáreo com 34 semanas.
Trigemelares Monocoriônicos ou dicoriônicos:
- 12 semanas: aconselhamento considerando opções de conduta expectante ou embriorredução.
- Ultrassom com 12 e 16 semanas e então a cada 2 semanas até o parto.
- Se houver discordância no peso fetal >15%, discordância de líquido amniótico ou Doppler anormal, reavaliar em 1 semana.
- Se não houver complicação, parto cesáreo com 32-34 semanas.
Monocoriônicos: gemelaridade imperfeita
Prevalência:
- 1% dos gêmeos Monocoriônicos. • Resultado de divisão incompleta da massa embrionária 12 após a fertilização.
Diagnóstico Ultrassonográfico:
- Gêmeos fundidos em gestação monocoriônica monoamniótica.
- Classificados de acordo com o local da junção seguido do sufixo pagos (do grego “o que está fixado”). Tipo mais comum é toracópagos (75%) com fusão no tórax e abdome e frequentemente coração, fígado e intestinos compartilhados.
- Outros tipos incluem pygopagos (fetos fundidos pelas nádegas), isquiópagos (fetos fundidos pela metade inferior do corpo, com colunas fundidas ponta com ponta em ângulo de 180 graus), craniópagos (crânios colados e corpos separados), onfalópagus (fusão de abdome inferior com fígado e intestinos compartilhados).
Anormalidades associadas:
- A incidência de anormalidades cromossômicas e síndromes genéticas não está aumentada.
Investigação:
- Ultrassonografia detalhada e avaliação por equipe multidisciplinar.
Manejo:
- Interrupção da gestação, morte intrauterina ou neonatal em > 90% dos casos.
- Se a gravidez continuar, parto deve ser cesáreo em centro com experiência no manejo de tais gestações.
Prognóstico:
- Altíssimo risco de deficiência nos sobreviventes.
Recorrência:
- Não há risco aumentado.
Monocoriônicos: morte de 1 gemelar
Prevalência:
- Morte espontânea de 1 gemelar ocorre em 1% dos gêmeos monocoriônicos.
Implicações:
- Morte de um gemelar é associada a hemorragia agudo do outro feto para a unidade feto-placentária do gemelar morto. Para o segundo feto, há 15% de risco para morte e ? 25% dos sobreviventes apresentam dano neurológico. Há também alto risco (60-70%) de parto prematuro.
- Se a morte de um feto ocorre < 24 semanas, é mais provável que o outro também venha a falecer, mas se sobreviver, dano neurológico pode ser menor; se a morte ocorre > 24 semanas, é mais provável que o segundo gêmeo sobrevivia, mas também mais provável que haja dano cerebral.
Manejo:
- Avaliação ultrassonográfica para estimar se a morte ocorreu nas últimas 48h ou antes (edema generalizado leve, ascite e derrame pleural) e avaliação do sobrevivente a procura de evidências de hemorragia cerebral, falência cardíaca e circulação hiperdinâmica com alto pico de velocidade sistólica na artéria cerebral média ( PVS ACM).
- Morte < 48h e PVS ACM ? 1.5MoM: transfusão intrauterina e repetição do Doppler 5-10h após. Se o PVS ainda estiver > 1.5MoM, outra transfusão é necessária. Se houver polidramnia e o comprimento cervical for < 20mm, considerar amniodrenagem.
- Morte < 48he PVS ACM < 1.5MoM: repetir o Doppler nas próximas 24h. Se o PVS se tornar > 1.5MoM, uma transfusão intrauterina se faz necessária. Se houver polidramnia e o comprimento cervical for < 20mm, considerar amniodrenagem. • Morte > 48h e nenhuma evidência de falência cardíaca fetal: conduta expectante independentemente do PVS ACM. Se houver polidramnia e o comprimento cervical for < 20mm, considerar amniodrenagem.
- Se o feto sobreviver: neurossonografia deve ser realizada em 2-4 semanas e RMN com 32 semanas para determinar se há evidência de dano cerebral. Se desenvolvimento cerebral, crescimento fetal e Dopplers forem normais, não há necessidade de adiantar o parto. Se houver evidência de dano cerebral, em alguns países, a interrupção da gestação é uma opção legal.
Monocoriônicos: monoamnióticos
Prevalência:
- 5% dos gêmeos monoamnióticos.
- Resultam da divisão da massa embrionária 9 dias após a fertilização.
Diagnóstico Ultrassonográfico:
- Ausência de membrana entre os gêmeos. Os dois cordões umbilicais tem inserções próximas na placenta com anastomoses de grande calibre entre as duas circulações fetais.
- Color Doppler demonstra entrelaçamento dos cordões na maioria dos casos o que ocorre geralmente desde o primeiro trimestre. Esse fato não contribui para morte fetal ou dano cerebral.
- O número de vesículas vitelínicas não prediz de forma acurada a amnionicidade, pois alguns gêmeos monoamnióticos têm 2 vesículas e poucos diamnióticos têm apenas uma.
Anormalidades associadas:
- A incidência de anomalias cromossômicas e síndromes genéticas não é aumentada.
- Risco de discordância de anomalias estruturais (20%) é maior que em gêmeos monocoriônicos diamnióticos (8%).
Investigação:
- Ultrassonografia detalhada.
- Ultrassom com 12 e 16 semanas e então a cada 2 semanas até o parto. Avaliação do crescimento, desenvolvimento cerebral, volume de líquido amniótico e índice de pulsatilidade da artéria umbilical, ACM e ducto venoso em ambos os fetos.
- Se houver discordância no tamanho dos fetos > 15% ou qualquer anormalidade no Doppler , revisar em 1 semana.
Manejo:
- Morte fetal, usualmente dos dois gêmeos, ocorre em 70% dos casos ( 50% < 20 semanas, 15% entre 20-32 semanas e 5% ? 32 semanas).
- A causa mais comum de morte fetal, que ocorre subitamente e de forma imprevisível, é exsanguinação acuda através das grandes anastomoses entre os dois cordões, deflagrada provavelmente por compressão.
- Discordância para anormalidade maior: uma opção é oclusão endoscópica do cordão do feto afetado, seguida de secção do cordão para evitar morte subsequente do gemelar saudável mais tarde na gestação.
- A síndrome de transfusão feto-fetal (STFF) ocorre raramente e pode ser suspeitada pelo desenvolvimento de polidramnia e o achado de bexiga pequena ou ausente no doador e grande no receptor. A proximidade dos cordões torna impossível a opção de ablação endoscópica a laser dos vasos comunicantes. Opções alternativas são amniodrenagem, adiantar o parto ou oclusão e secção endoscópica do cordão de um dos gêmeos.
- Não há necessidade para hospitalização. Parto deve ser através de cesariana eletiva com 32 semanas.
Recorrência:
- Não há risco aumentado.
Monocoriônicos: crescimento restrito seletivo
Prevalência:
10-15% dos gêmeos monocoriônicos.
Diagnóstico Ultrassonográfico:
- Peso fetal estimado abaixo do percentil 5 no feto pequeno e ? 25% de discordância entre os fetos.
- O líquido amniótico no feto menor está reduzido e normal do outro gemelar.
- A condição é subdividida me 3 tipos de acordo com o fluxo diastólico da artéria umbilical do menor feto ao Doppler:
- Tipo I: diástole positiva.
- Tipo II: diástole ausente ou reversa.
- Tipo III: diástole cíclica alternando de positiva para ausente e reversa.
- Se, na presença de discordância ? 25% entre os pesos estimados dos fetos, houver polidramnia no cavidade do maior feto, a condição é crescimento restrito seletivo com STFF sobreposta.
Anormalidades associadas:
- A incidência de anormalidades cromossômicas e síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada.
- Ultrassom semanal para monitorar crescimento, volume de líquido e índice de pulsatilidade na artéria umbilical, ACM e ducto venoso.
Manejo:
- Tipos I e II com STFF: ablação endoscópica a laser dos vasos placentários comunicantes.
- Tipo I sem STFF: conduta expectante com monitoramento rigoroso para definir melhor momento para parto. Se os achados ao Doppler permanecerem normais, cesariana eletiva entre 34-35 semanas. Há sobrevivência intacta de ambos os gêmeos em 95% dos casos.
- Tipo II sem STFF: há alto risco de morte perinatal e deficiência para ambos os fetos.
- ?26 semanas: a melhor conduta é expectante com monitoramento rigoroso e parto caso a diástole do ducto venoso se torne negativa ou reversa.
- <26 semanas: a melhor conduta é ablação endoscópica a laser dos vasos comunicantes placentários. Sobrevivência do feto maior é 70% e do menor depende da diástole do ducto venoso: 40% se positiva, 10% se negativa ou reversa. O risco de lesões cerebrais no neonato depende primariamente da idade gestacional ao nascimento e varia de 20% para nascimento < 26 semanas para 5% para nascidos ?32 semanas. Uma conduta alternativa é oclusão do cordão do feto menor; a sobrevivência do feto maior é 90%.
- Tipo III: os dois cordões são adjacentes e a gestação se comporta como monoamniótica; desenvolvimento de STFF é raro e morte súbita inesperada pode ocorrer em 20-30% dos casos. Cirurgia a laser pode ser impossível e a melhor conduta é monitoramento rigoroso e parto a depender da diástole do ducto venoso do feto pequeno. Se a diástole do ducto é positiva, parto cesáreo eletivo deve ser feito com 32 semanas. Há 20% de risco de lesão cerebral neonatal, sendo esse mais elevado no feto maior do que no menor.
Recorrência:
- Não há risco aumentado.
Monocoriônicos: sequência anemia-policitemia
Prevalência:
- Espontânea: 5% dos monocoriônicos. Usualmente ocorre > 26 semanas.
- Após ablação a laser de vasos placentários: 2-10% dos monocoriônicos.
Diagnóstico Ultrassonográfico:
- TAPS ocorre devido à transfusão sanguínea lenta do doador para o receptor por algumas poucas e pequenas anastomoses vasculares arteriovenosas levando a grande discordância nos níveis de hemoglobina.
- Não há diferença substancial entre o tamanho e volume de líquido dos gêmeos.
- A placenta do feto anêmico é espessa e hiperecóica, enquanto a do policitêmico é fina e translúcida.
- O PVS ACM do feto anêmico é aumentado ( .1.5MoM), enquanto o do policitêmico é diminuído (<1MoM).
- O feto an6emico pode ter coração dilatado, regurgitação tricúspide e ascite. O fígado do policitêmico tem aspecto estrelado devido a diminuição da ecogenicidade do seu parênquima e brilho aumentado da parede dos vasos portais.
Anormalidades associadas:
- A incidência de anormalidades cromossômicas e síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada, incluindo ecocardiograma (para avaliar função cardíaca) e medida do PVS ACM (para predizer grau de anemia e policitemia).
Manejo:
- <26 semanas: ablação endoscópica a laser de vasos placentários comunicantes. Subsequentemente, ultrassom semanal para monitorar crescimento, anatomia cerebral e PVS ACM. RMN cerebral ?32 semanas deve ser considerada para diagnóstico de desordens de migração neuronal. Se ambos os fetos estiverem se desenvolvendo normalmente, parto vaginal pode ser feito com 37 semanas.
- 26-30 semanas: transfusões intrauterinas para o feto anêmico e exsanguineotransfusão com solução Hartman para o policitêmico. Avaliação com Doppler a cada 2-3 dias pois pode ser necessária intervenção a cada 3-4 dias. Parto cesáreo com 30-32 semanas.
- >30 semanas: parto cesáreo.
Prognóstico:
- Retardo no neurodesenvolvimento em até 20% dos casos o que depende principalmente da idade gestacional ao nascimento. O risco é maior para o feto receptor.
Recorrência:
- Não há risco aumentado.
Monocoriônicos: sequência TRAP
Prevalência:
- 2-3% dos monocoriônicos.
Diagnóstico Ultrassonográfico:
- Gêmeos monocoriônicos com um feto normal ( feto bomba) e outro sem atividade cardíaca ( raramente, um coração rudimentar pode ter pulsações lentas) e graus variáveis de déficit de desenvolvimento da cabeça e membros superiores e hidropsia ( feto receptor).
- Color Doppler no feto receptor demonstra fluxo pulsátil reverso de uma anastomose arterio-arterial umbilical e retorno venoso para o feto bomba via anastomose veno-venosa. Ocasionalmente, o cordão do feto acárdico surge diretamente do cordão do feto bomba.
- O tamanho da massa acárdica, é de valor prognóstico para a sobrevivência do feto bomba.
- Cerca de 50% dos fetos bomba, morrem antes ou depois do nascimento de insuficiência cardíaca congestiva ou parto extremamente prematuro por polidramnia.
Anormalidades associadas:
- A incidência de anormalidades cromossômicas e síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada, incluindo ecocardiograma para avaliar função cardíaca do feto bomba.
Terapia Fetal:
- Tratamento pré-natal é realizado através da oclusão do fluxo sanguíneo do gêmeo acárdico. Diversos métodos foram usados, incluindo ablação dos vasos do cordão umbilical por laser ou diatermia, coagulação a laser das anastomoses placentárias ou ablação dos vasos interfetais por diatermia monopolar, laser ou radiofrequência. Quando esses métodos são usados entre 16-18 semanas, a taxa de sobrevivência do feto bomba é de 80%.
- O manejo de escolha é coagulação a laser ou radiofrequência guiados por ultrassom dos vasos do cordão umbilical dentro do abdome do feto acárdico entre 11 e 13 semanas. A sobrevivência é de 70-75% que é menor que os 80% alcançados com a intervenção entre 16-18 semanas.
- Entretanto, atraso na intervenção entre o diagnóstico de sequência TRAP entre 11-13 semanas até 16-18 semanas é associado com interrupção espontânea do fluxo do feto acárdico em 60% dos casos e em cerca de 50% desses ocorre morte ou dano cerebral do feto bomba.
Seguimento:
- Intervenção intrauterina: ultrassom em 1 semanas para confirmar que o feto bomba está vivo e que não há fluxo no feto acárdico. Subsequentemente, acompanhamento rotineiro.
- Sem intervenção intrauterina: ultrassom a cada 2-3 semanas para monitorar crescimento do feto acárdico e função cardíaca e líquido amniótico do feto bomba.
Parto:
- Assistência obstétrica e parto de rotina.
Prognóstico:
- Depende da idade gestacional ao nascimento.
Recorrência:
- Não há risco aumentado.
Monocoriônicos: síndrome de transfusão feto-fetal
Prevalência:
- 10-15% dos monocoriônicos.
Diagnóstico Ultrassonográfico:
- Há desequilíbrio na rede de fluxo sanguíneo das comunicações vasculares placentárias de um feto, o doador, para o outro, o receptor.
- 11-14 semanas: STFF precoce é suspeitada na presença de discordância no tamanho das cavidades amnióticas, discordância ? 20% na espessura da translucência nucal (TN) de cada feto ou diástole zero ou reversa no ducto venoso, usualmente no feto com maior TN.
- ? 15 semanas: oligodramnia ( maior bolsão vertical ? 2cm) na cavidade do feto doador oligúrico ou anúrico e polidramnia (? 6cm com 15-17 semanas, ? 8cm com 18-20 semanas e > 10 cm ? 20 semanas) na cavidade do receptor poliúrico.
- A condição é subdividida em 4 estágios de acordo com o fluxo diastólico da artéria umbilical e ducto venoso ao Doppler:
- Estágio 1: bexiga do doador visível, diástole positiva nos dois vasos de ambos os fetos.
- Estágio 2: bexiga do doador não visível, diástole positiva nos dois vasos de ambos os fetos.
- Estágio 3: diástole ausente ou reversa em qualquer um dos vasos de qualquer feto.
- Estágio 4: presença de ascite ou hidropsia fetal; usualmente no receptor.
Anormalidades associadas:
- A incidência de anormalidades cromossômicas e síndromes genéticas não é aumentada.
Investigação:
- Ultrassonografia detalhada.
- Ultrassom semanal para monitorar crescimento fetal, líquido amniótico e índice de pulsatilidade na artéria umbilical, ACM e ducto venoso de ambos os fetos.
Manejo:
- Discordância de líquido amniótico ( mas não o suficiente para preencher critérios de sequência oligodramnia/polidramnia) com Doppler normal:
- Sobrevivência geral: 95%.
- Progressão para STFF: 15%.
- Ultrassom a cada 1-2 semanas para monitorar evolução.
- Estágio 1:
- Sobrevivência geral 85%, pelo menos um gemelar 90%.
- Ultrassom semanal para monitorar evolução.
- Ablação endoscópica a laser de vasos comunicantes placentários se progressão para estágios 2-4 ou polidramnia crescente com encurtamento cervical.
- Estágios 2-4:
- <28 semanas: a melhor conduta é ablação endoscópica a laser dos vasos comunicantes placentários; ablação de todos os vasos comunicantes e a área entre eles também deve ser coagulada para atingir completa dicorionização da placenta.
- ?28 semanas: a melhor opção é parto cesáreo e o momento é definido pelos achados ao Doppler de artéria umbilical e ducto venoso de ambos os fetos.
- Estágio 2: sobrevivência geral 75%, pelo menos 1 gemelar 85%.
- Estágio 3 e 4: sobrevivência geral 60-70%, pelo menos um gemelar 75-85%.
- Déficit no neurodesenvolvimento nos sobreviventes: 5-10%.
- Seguimento após terapia com laser: ultrassom com Doppler semanal até resolução dos sinais de STFF e normalização dos achados ao Doppler e a cada 2 semanas após, com atenção especial ao sinais de dano cerebral, recorrência de STFF e desenvolvimento de TAPS.
- Normalização do líquido amniótico ocorre após 1 semana. Resolução da disfunção cardíaca no receptor e da hidropsia no estágio 4, usualmente ocorre após 3-4 semanas.
- Em cerca de 1 em 1.000 casos, pode haver amputação de membro devido a evento trombótico ou banda amniótica.
Parto:
- Vaginal com 37 semanas se houver crescimento e Doppler normal em ambos os fetos.
Recorrência:
- Não há risco aumentado.
Anormalidades estruturais
Dicoriônicos:
- A prevalência de defeitos por feto é a mesma que em fetos únicos (2%), logo o risco de defeito em pelo menos um feto é 2 vezes maior (4%) que na gestação única. Em 10% dos casos, ambos os fetos são afetados (concordância) e em 90% apenas um é afetado (discordância).
Monocoriônicos:
- A prevalência de defeitos por feto é 2 vezes maior que em fetos únicos, logo o risco de ao menos um gemelar afetado é 4 vezes maior (8%) do que o da gestação única. Em 20% ambos são afetados (concordância) e em 80% apenas um (discordância).
Manejo de gestações com anormalidades discordantes:
- Essas gestações pode ser conduzidas com conduta expectante ou feticídio seletivo do gêmeo anormal.
- Em casos em que a anormalidade é não letal mas pode resultar em grave deficiência, os pais precisam decidir se o ônus potencial de um filho deficiente é suficiente para arriscar perder o gemelar normal devido a complicações relacionadas ao feticídio.
- Em casos em que a anormalidade é letal, é melhor evitar riscos associados ao feticídio seletivo, a não ser que a condição por si só ameace a vida do gemelar normal. Por exemplo, em anencefalia ou trissomia 18 (com atresia esofagiana ou hérnia diafragmática associada), há risco > 50% de desenvolvimento de polidramnia entre 24-26 semanas, colocando o feto normal em risco de parto prematuro e morbimortalidade associada.
Feticídio seletivo em dicoriônicos:
- Feticídio pode ser realizado pela injeção intracardíaca de cloreto de potássio.
- Feticídio entre 11-14 semanas: risco de abortamento 7% e risco de parto <32 semanas 6%.
- Feticídio ?16 semanas: risco de abortamento 14% e risco de parto <32 semanas 20%.
- Feticídio com 32 semanas: é opção legal em alguns países para evitar risco de abortamento e parto prematuro extremo.
Feticídio seletivo em monocoriônicos:
- Pode ser realizado pela oclusão do cordão umbilical através de laser endoscópico, fórceps bipolar guiado por ultrassom ou radiofrequência.
- Feticídio ?16 semanas: risco de abortamento 20% e risco de parto <32 semanas 20%.
Gestação trigemelar
Opções de conduta entre 11-13 semanas:
- Em gestações Trigemelares diagnosticadas durante o primeiro trimestre, as opções de conduta incluem continuar com gravidez ou embriorredução (ER) para gemelares ou gestação única. • A consequência da ER é diminuição da taxa de parto prematuro extremo, mas aumento da taxa de abortamento <24 semanas (tabela abaixo).
- Trigemelares tricoriônicos (TC): ER é realizada através da injeção intracardíaca de cloreto de potássio (KCl).
- Trigemelares dicoriônicos (DC): ER com KCl envolve ou o feto separado ou ambos os monocoriônicos (MC); o KCl injetado apenas em um gemelar MC pode ser transferido para seu par através de anastomoses placentárias ou a morte de um pode levar a hemorragia do seu par para a unidade fetoplacentária morta com consequente morte ou déficit de neurodesenvolvimento do sobrevivente.
- Em DC, comparado a TC, há maior risco de abortamento tanto para conduta expectante quanto para ER para MC, fato que pode ser atribuído em parte ao desenvolvimento subsequente de restrição seletiva de crescimento e STFF.
- Outra opção para trigemelares DC é a ER para gestação gemelar DC através de ablação a laser guiada por ultrassom dos vasos pélvicos de um do fetos MC. Os riscos de abortamento e parto prematuro extremo são menores com esse método do que com outras opções. Entretanto, com laser intrafetal, metade das gestações vai resultar no nascimento de um e não dois bebês, pois o outro gemelar MC morre nas 2 semanas seguintes ao procedimento.
| Opções de conduta 11-13 semanas | Aborto 12+0 a 23+6 sem |
Parto Prematuro 24+0 a 32+6 sem |
| Trigemelar tricoriônico | ||
| – Conduta expectante | 3% | 35% |
| – Redução p/DC por KCl | 7% | 13% |
| – Redução p/ feto único por KCl | 10% | 8% |
| Trigemelar dicoriônico | ||
| – Conduta expectante | 9% | 38% |
| – Redução p/ MC por KCl | 13% | 23% |
| – Redução p/ feto único por KCl | 18% | 9% |
| – Redução p/ DC por laser | 3% | 7% |
Síndrome de transfusão feto-fetal:
- Em gestação trigemelar complicada por STFF grave, a melhor opção de conduta é ablação endoscópica a laser dos vasos comunicantes placentários.
- Trigemelar DC: o feto separado pode impor dificuldade técnica na escolha do local de entrada apropriado para o fetoscópio mas em geral esse problema pode ser facilmente superado. Consequentemente, o desfecho de gestações afetadas é similar àquele de gêmeos MC mas com risco inevitavelmente maior de parto prematuro extremo.
- Sobrevivência : geral 75%, pelo menos um feto 95%. Nascimento < 32 semanas: 55%.
- Trigemelar MC: cirurgia endoscópica a laser pode ser tecnicamente difícil pela necessidade de ablação dos vasos comunicantes entre todos os 3 fetos. O endoscópio é introduzido na cavidade amniótica do feto receptor e laser é usado para coagula os vasos entre esse feto e cada um dos outros 2. Subsequentemente, o fetoscópio avança entre através da membrana interfetal para a cavidade de um dos outros fetos para coagular as conexões vasculares entre eles. Como resultado de problemas técnicos ou da inabilidade de atingir o objetivo de completa separação entre todos os fetos o desfecho é pior do que o de trigemelares dicoriônicos.
- Sobrevivência: geral 55%, pelo menos um feto 80%. Parto < 32 semanas: 65%.
Agradecimentos
Diretor: Kypros Nicolaides
Coleta e edição de vídeos: Jader Cruz, Nobuhiko Hayashi
Principal contribuinte de vídeos: Francisca Molina
Texto: Magdalena Litwi?ska
Programação: Dimitris Vichos
Tradução: Carolina Ribeiro
Rastreio de Pré-eclâmpsia
Bem vindo a este curso educacional sobre rastreio de pré-eclâmpsia (PE) no primeiro, segundo e terceiro trimestres da gestação.
Este curso abordará::
- Histórico da PE, incluindo definição, prevalência, complicações e patogênese.
- Prevenção de PE, por medidas como repouso no leito, hábitos alimentares e uso de baixa dose de aspirina.
- Predição de PE por uma combinação de história e características maternas com marcadores bioquímicos e biofísicos.
- Desempenho do teste para rastreio de PE no primeiro, segundo e terceiros trimestres da gravidez.
- Estratificação do manejo na gravidez.
O rastreio entre 11-13 semanas identifica um grupo de alto risco, no qual a administração de aspirina pode reduzir substancialmente a incidência de pré-eclâmpsia pré-termo.
O rastreio durante o segundo e terceiro trimestres identifica um grupo de alto risco, no qual o monitoramento intensivo pode levar ao diagnóstico precoce de pré-eclâmpsia e redução dos eventos perinatais adversos associados à doença.
O curso dura cerca de 1 hora, mas não precisa ser finalizado em uma única sessão.
Ao completar o curso, você obterá um certificado de conclusão.
O curso é obrigatório como parte da certificação da FMF em Rastreio de Pré-eclâmpsia.
Conteúdos do curso
Antecedentes para a pré-eclâmpsia
Definição, prevalência, implicações, patogênese
Prevenção da pré-eclâmpsia
Repouso no leito e manipulações dietéticas
Aspirina em dose baixa
Predição de pré-eclâmpsia
Características maternas e história
Marcadores bioquímicos: PLGF, sFLT-1, PAPP-A
Marcadores biofísicos: MAP, UTPI
Desempenho da triagem
Desempenho em 11-13 semanas
Desempenho em 20-24 semanas
Desempenho em 30-33 semanas
Desempenho em 35-37 semanas
Estratificação do manejo da gravidez
Histórico da pré-eclâmpsia
Definição
Pré-eclâmpsia (PE) é uma síndrome multissistêmica que surge durante a segunda metade da gestação. É caracterizada por hipertensão e proteinúria ou acometimento de orgãos-alvo maternos.
Hipertensão é pressão arterial sistólica ?140 mmHg e/ou pressão diastólica ?90 mmHg em ?2 ocasiões com intervalo de 4 horas, com aparecimento após 20 semanas de gestação em mulheres previamente normotensas.
Proteinúria é a presença de ?300 mg de proteína em uma amostra de urina de 24 horas ou a razão entre proteína/creatinina urinária ?30 mg/mmol ou dois resultados de pelo menos ++ de proteína na análise por fita de uma amostra de jato médio de urina ou por catéter.
Em PE superajuntada à hipertensão crônica (história de hipertensão antes da concepção ou presença de hipertensão antes de 20 semanas de gestação), proteinúria ou acometimento de orgãos-alvo maternos devem ocorrer após 20 semanas de gestação.
Acometimento de órgãos-alvo maternos é definido pelo o aparecimento de quaisquer um dos abaixo:
- Insuficiência renal – creatinina sérica ?90 ?mol/L.
- Disfunção hepática – transaminases hepáticas em altos níveis séricos (?2 vezes o limite de normalidade) e/ ou dor abdominal supra-umbilical intensa e persistente irresponsiva à medicação.
- Complicações neurológicas – eclâmpsia, acidente vascular cerebral, confusão mental, hiperreflexia acompanhada de clonus, cefaléia intensa acompanhada por hiperreflexia, amaurose ou escotomas visuais persistentes.
- Complicações hematológicas – contagem plaquetária <150,000/dL, coagulação intravascular disseminada (CIVD) ou hemólise.
Prevalência
A PE ocorre em 2-5% das gestações. A taxa depende das características demográficas da população sob análise. Por exemplo, em mulheres negras a taxa é 2-3 vezes maior do que em mulheres brancas.
Em um terço dos casos essa condição resulta no parto <37 semanas de gestação (PE pré-termo) e em dois terços o parto ocorre ?37 semanas (PE a termo).
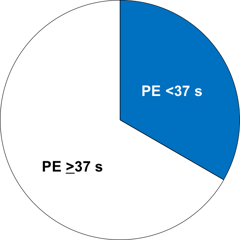
Complicações
A PE é uma das principais causas de morbimortalidade materna e perinatal e é responsável por mais de 50 mil mortes maternas por ano em todo o mundo. As complicações mais graves, que podem levar à morte materna, são a eclâmpsia (convulsões ou coma em uma mulher com PE), hemorragia cerebral ou acidente vascular cerebral, coagulação intravascular disseminada (CIVD) e síndrome HELLP (hemólise, enzimas hepáticas elevadas e plaquetas baixas). Outras complicações graves incluem edema cerebral, amaurose, insuficiência renal, insuficiência hepática ou edema pulmonar.
A PE está associada à redução do suprimento sanguíneo placentário, com consequente comprometimento do crescimento fetal, oxigenação e aumento do risco de morte fetal. Além disso, o parto prematuro ocorre em uma grande parte das mulheres com PE, por indicações maternas e / ou fetal e, portanto, os bebês estão sujeitos aos riscos adicionais decorrentes da prematuridade. Estes incluem morte neonatal, hemorragia cerebral, convulsões, dificuldades respiratórias e de alimentação, icterícia, retinopatia e hospitalização prolongada. A PE e a eclâmpsia são responsáveis por cerca de 25% dos óbitos intrauterinos e mortes neonatais e por 15% dos neonatos com baixo peso ao nascimento.
O risco de desfecho adverso para a mãe e feto / bebê é muito maior na ocorrência de PE pré-termo do que na PE a termo.
Complicações a longo prazo
Mulheres que desenvolvem PE, em comparação com aquelas que não desenvolvem PE, têm o dobro do risco de sofrer de doenças cardiovasculares (DCV) ao longo da vida, incluindo hipertensão, doença cardíaca isquêmica, AVC e morte por DCV. É incerto se esta associação é porque a PE causa dano vascular que, a longo prazo, leva a DCV ou, mais provável, que as mulheres destinadas a desenvolver DCV sejam estimuladas pela gravidez a desenvolver PE.
Crianças expostas à PE antes do nascimento, em comparação com as que nascem após uma gravidez normal, têm o dobro de risco de sofrer de paralisia cerebral e este risco é mediado pela ocorrência de parto prematuro, restrição de crescimento ou ambos. Da mesma forma, crianças e jovens adultos expostos à PE têm uma maior pressão arterial e índice de massa corporal em relação aqueles nascidos após uma gravidez normal e apresentam um risco aumentado para desenvolver DCV e diabetes na vida adulta.
Patogênese
As principais responsáveis pela irrigação do útero são as artérias uterinas esquerda e direita, que se ramificam para formar, em sequência: as artérias arqueadas, radiais, basais e espiraladas.
Na gravidez, o blastocisto se implanta no endométrio materno. A camada externa do blastocisto se desenvolve em trofoblasto, que se diferencia em vilos trofoblásticos, que darão origem às vilosidades coriônicas, responsáveis por transportar nutrientes e oxigênio entre o feto e a mãe; e os trofoblastos extravilosos, que invadem e transformam as artérias espiraladas. Essencialmente, os trofoblastos substituem o revestimento endotelial e destroem o tecido musculoelástico das paredes das artérias espiraladas para que sejam convertidas então, de vasos musculares estreitos e tortuosos, em grandes canais não musculares, aumentando assim o fluxo sanguíneo materno para a placenta.
Este processo fisiológico ocorre em duas etapas: a primeira onda de invasão trofoblástica envolve as artérias espiraladas na decídua (endométrio da gravidez) e começa às 8 semanas de gestação, enquanto a segunda envolve as artérias espiraladas no terço interno do miométrio e ocorre às 14-18 semanas.
Na PE, particularmente na PE pré-termo, o processo fisiológico de placentação é prejudicado. Há invasão trofoblástica em 50-70% das artérias espiraladas; no entanto, isso não se estende aos segmentos miometriais e é confinado à parte decidual dos vasos. As artérias espiraladas são menos dilatadas do que as artérias normalmente transformadas e o fornecimento de sangue à placenta é reduzido. A razão pela qual, em algumas gestações, ocorre uma falha na placentação ainda é desconhecida, mas causas genéticas e imunológicas podem estar envolvidas.
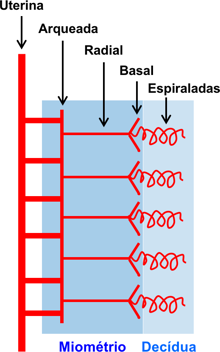
Patogênese
A redução da perfusão placentária provoca o estresse oxidativo que, por sua vez, desencadeia a liberação de fatores derivados do trofoblasto que entram na circulação materna, causando danos nas células endoteliais do rim, fígado, cérebro e placenta e uma resposta inflamatória exacerbada, que são a base de muitas das mudanças observadas na PE.
Os fatores placentários liberados em resposta ao estresse incluem a proteína anti-angiogênica (sFLT1), que está aumentada na PE. Já a concentração circulante do fator de crescimento angiogênico placentário (PLGF) está reduzida na PE. Este desequilíbrio angiogênico resulta no aumento da inflamação vascular materna e disfunção endotelial generalizada.
Ao contrário da PE pré-termo, que é caracterizada por placentação deficiente, a placenta da PE a termo é em geral normal. Em mulheres com comorbidades, tais como hipertensão crônica, a disfunção endotelial está presente mesmo antes da gravidez. Nesses casos, a PE pode se desenvolver mesmo na ausência ou com menor grau de placentação deficiente; a disfunção endotelial pré-existente é ainda mais exacerbada pelas alterações fisiológicas da gravidez, uma vez que as gestações normais possuem uma resposta inflamatória sistêmica de menor intensidade.
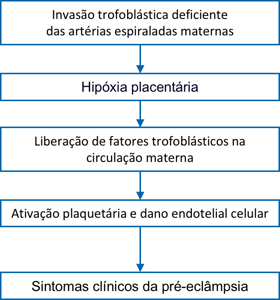
Prevenção da pré-eclâmpsia
Repouso no leito e hábitos alimentares
A taxa de PE não é reduzida pelo repouso no leito, restrição de atividade física ou hábitos alimentares, incluindo restrição de ingestão de sal ou suplementação alimentar com magnésio, zinco, folato, vitaminas C, D e E ou óleo de peixe.
A suplementação dietética com cálcio em mulheres com dietas pobres neste mineral pode diminuir pela metade a taxa de PE. Dados preliminares sugerem também que o uso profilático de paravastatinas pode beneficiar mulheres com alto risco de PE.
Baixa dose de aspirina
O uso profilático de baixa dose de aspirina na prevenção de PE tem sido amplamente estudado. Uma meta-análise de ensaios clínicos mostrou que a administração de baixas doses de aspirina em gestações de alto risco resultou em uma incidência 10% menor de PE. No entanto, na maioria dos estudos, a aspirina foi iniciada após 16 semanas de gestação e a uma dose menor que 100 mg/dia.
Em contraste, outras meta-análises mostraram que a aspirina iniciada antes de 16 semanas reduziu pela metade a taxa de PE, enquanto que a aspirina iniciada após 16 semanas não mostrou um benefício significativo. Além disso, o efeito benéfico da aspirina era dependente da dose, com uma maior redução na incidência de PE associada a uma dose maior que 100 mg/dia.
- Hofmeyr GJ, Lawrie TA, Atallah AN, Duley L, Torloni MR. Calcium supplementation during pregnancy for preventing hypertensive disorders and related problems. Cochrane Database Syst Rev 2014;6: CD001059.
- Askie LM, Duley L, Henderson-Smart DJ, Stewart LA. Antiplatelet agents for prevention of preeclampsia: a meta-analysis of individual patient data. Lancet 2007; 369: 1791-8.
- Roberge S, Nicolaides KH, Demers S, Villa P, Bujold E. Prevention of perinatal death and adverse perinatal outcome using low-dose aspirin: a meta-analysis. Ultrasound Obstet Gynecol 2013; 41: 491-9.
- Crandon AJ, Isherwood DM. Effect of aspirin on incidence of pre-eclampsia. Lancet 1979; 1: 1356.

Em 1543 AC Os egípcios usavam extratos do salgueiro para alívio da dor.
Em 400 AC Hipócrates usou um pó feito da casca e folhas do salgueiro para tratar cefaléia, dores e febre.
Em 1828 Johann Buchner na Universidade de Munique, extraiu o ingrediente ativo da planta do salgueiro e o nomeou de salicilato (termo em Latin para salgueiro).
Em 1915 Bayer desenvolveu comprimidos de aspirina.
Em 1979 Crandon e Isherwood relataram que as mulheres que haviam tomado aspirina regularmente durante a gravidez apresentaram menor probabilidade de desenvolver PE do que as mulheres que não a tinham tomado.
O estudo ASPRE
O estudo ASPRE (Combined Multimarker Screening and Randomized Patient Treatment with Aspirin for Evidence-Based Preeclampsia Prevention) foi um estudo multicêntrico internacional. O rastreio de rotina para PE pré-termo foi realizado em cerca de 27.000 gestações únicas, entre 11-13 semanas de gestação, utilizando o algoritmo da FMF que combina fatores maternos e biomarcadores.
As mulheres elegíveis (n = 1.620) com risco estimado de PE pré-termo >1 em cada 100, que concordaram em participar do estudo, foram alocadas aleatoriamente para aspirina (150 mg/dia) ou placebo, de 11-14 semanas de gestação até 36 semanas. As participantes foram instruídas a tomar o comprimido à noite, ao invés de dia, porque há evidências de que o tratamento neste período possa ser superior na redução da taxa de PE.
O uso da aspirina foi associado a uma redução de 62% na incidência de PE pré-termo e redução de 82% na incidência de PE antes de 34 semanas de gestação.
Análise secundária dos dados do estudo ASPRE mostrou que:
- O efeito benéfico da aspirina depende da adesão e a redução da incidência de PE pré-termo pode ser ao redor de 75% naquelas com comprometimento maior que 90% e de apenas 40% naquelas com adesão menor que 90%.
- Na hipertensão crônica, a aspirina pode não ser útil na prevenção de PE pré-termo.
- Consequentemente, se as participantes com hipertensão crônica tivessem sido excluídas do estudo e a adesão tivesse sido ?90%, a aspirina poderia ter potencialmente reduzido a incidência de PE pré-termo em 95%.
Predição de pré-eclâmpsia
Método de rastreio da FMF
A abordagem da FMF para rastreio de PE é usar o teorema de Bayes, que combina o risco a priori de características maternas e histórico médico com os resultados de várias combinações de medidas biofísicas e bioquímicas feitas em diferentes momentos da gravidez.
Esta abordagem pressupõe que, se a gravidez continuasse indefinidamente, todas as mulheres desenvolveriam PE e o diagnóstico desta condição dependeria exclusivamente do momento da ocorrência do parto.
Os efeitos das variáveis características maternas, história e biomarcadores são modificar a distribuição média da idade gestacional ao parto na presença de PE, de modo que:
- Em gestações de baixo risco a distribuição da idade gestacional é deslocada para a direita com a implicação de que, na maioria das gestações, o parto ocorrerá antes do desenvolvimento de PE.
- Em gestações de alto risco a distribuição é deslocada para a esquerda e quanto menor for a idade gestacional média, maior será o risco de PE.
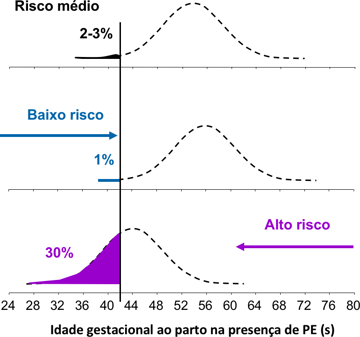
Características maternas e história
As variáveis, características maternas e história médica e obstétrica, que aumentam o risco de PE e, portanto, alteram a distribuição Gaussiana da idade gestacional com PE ao parto para a esquerda, incluem o aumento da idade materna, aumento do peso, origem racial Afro-Caribenha e do sul da Ásia, antecedentes médicos de hipertensão crônica, diabetes mellitus e lúpus eritematoso sistêmico ou síndrome antifosfolípide, concepção por fertilização in vitro e história familiar ou pessoal de PE. O risco de PE em mulheres grávidas pela primeira vez é três vezes maior do que em mulheres com gravidez anterior não complicada por PE. As mulheres que tiveram PE na primeira gravidez são até 10 vezes mais propensas a desenvolver PE em uma segunda gravidez.
O risco de PE é menor em mulheres altas do que em mulheres baixas e é reduzido em multíparas sem antecedente de PE. O efeito protetor contra PE, exercido por uma gravidez sem PE, diminui com o intervalo de tempo entre a gestação anterior e a gestação atual, de modo que após 15 anos o risco de PE é aproximadamente o mesmo que em mulheres nulíparas.
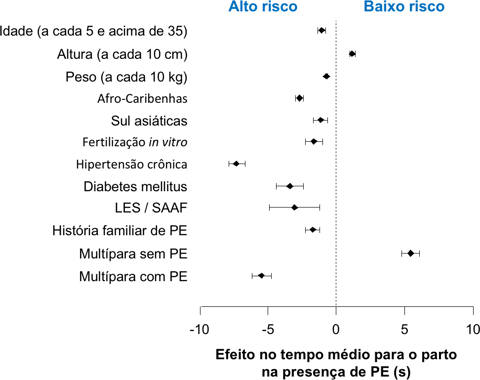
Medida da pressão arterial média (PAM)
A pressão sanguínea deve ser tomada por aparelhos automatizados autorizados, calibrados em intervalos regulares. Os esfigmomanômetros de mercúrio não devem ser usados devido a preocupações quanto ao desempenho clínico e à segurança destes instrumentos.
As medidas devem ser feitas por profissionais de saúde que tenham recebido treinamento adequado sobre o uso desses aparelhos.
As mulheres devem estar sentadas e suas pernas não devem estar cruzadas. O cruzamento das pernas pode aumentar da PAM.
Os braços da paciente devem ser apoiados ao nível do coração. Se a parte superior do braço estiver abaixo do nível do átrio direito, a PAM é superestimada e, se o braço estiver acima do nível do coração, a PAM é subestimada. Se o braço é mantido sem suporte, a PAM é superestimada.
O manguito adulto normal (22-32 cm) ou grande (33-42 cm) deve ser usado dependendo da circunferência do braço da paciente. Se o manguito for muito grande, a PAM é subestimada e, se o manguito for muito pequeno, a PAM é superestimada.
Após o repouso durante cinco minutos, duas medidas de PAM devem ser feitas em cada braço simultaneamente e a média das quatro deve ser considerada para avaliação do risco.
Medida da pressão arterial média (PAM)
Na predição de PE, a medida da PAM é mais útil do que a medida da pressão arterial sistólica e diastólica. A PAM é definida como a pressão arterial média durante um único ciclo cardíaco e é calculada a partir da seguinte fórmula: PAM = 2/3 pressão arterial diastólica + 1/3 pressão arterial sistólica.
Em gestaç?es normais, a PAM é dependente de fatores maternos, sendo os mais importantes o peso materno e hipertensão crônica. Consequentemente, para o seu uso efetivo no rastreio, essas co-variáveis precisam ser levadas em consideração, padronizando os níveis de PAM em valores normais de múltiplos da mediana (MoM).
Em gestaç?es que desenvolveram PE, a PAM está aumentada e a diferença nos valores normais de MoM é maior em idades gestacionais precoces do que nas avançadas, quando o parto por conta da PE torna-se necessário. Consequentemente, o desempenho do rastreio é superior para PE pré-termo do que para PE a termo.
A inclinação das linhas de regressão dos MoM da PAM em comparação com a idade gestacional ao parto, em gestações que desenvolvem PE, é maior conforme a idade gestacional. Consequentemente, o desempenho do rastreio melhora com o avanço da gravidez.
- Wright A, Wright D, Ispas CA, Poon LC, Nicolaides KH. Mean arterial pressure in the three trimesters of pregnancy: effects of maternal characteristics and medical history. Ultrasound Obstet Gynecol 2015; 45: 698-706.
- Tayyar A, Krithinakis K, Wright A, Wright D, Nicolaides KH. Mean arterial pressure at 12, 22, 32 and 36 weeks’ gestation in screening for pre-eclampsia. Ultrasound Obstet Gynecol 2016; 47: 573-9.
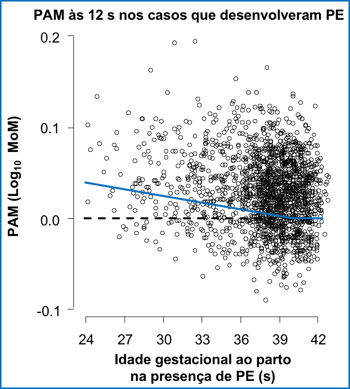
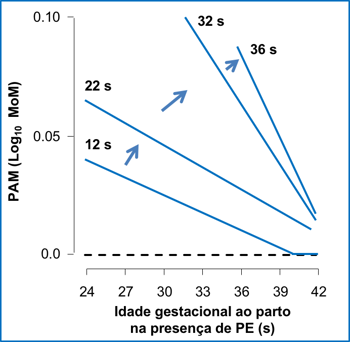
Medida do índice de pulsatilidade da artéria uterina (UTPI)
O UTPI pode ser medido por ultrasssonografia transabdominal ou transvaginal.
Na varredura transabdominal, no primeiro trimestre, deve-se obter um corte sagital do útero, identificando-se o canal endocervical e o orifício interno. O transdutor deve ser suavemente inclinado de lado a lado e o Doppler colorido deve ser usado para identificar cada artéria uterina ao longo da lateral do canal endocervical e útero, ao nível do orifício interno.
- No segundo e terceiro trimestres, o Doppler colorido deve ser usado para identificar cada artéria uterina no aparente cruzamento com as artérias ilíacas externas.
Na varredura transvaginal, as mulheres devem ser convidadas a esvaziar suas bexigas e colocadas na posição de litotomia dorsal. A sonda de ultrassom deve ser inserida então na vagina, e colocada, por sua vez, no fórnice lateral esquerdo e direito. As artérias uterinas são identificadas usando Doppler colorido ao nível do orifício interno do colo do útero.
Após a identificação de cada artéria uterina, o Doppler de onda pulsátil deve ser usado com a amostra configurada a 2 mm para cobrir todo o vaso. Deve-se ter cuidado para garantir que o ângulo de insonação seja inferior a 30º. É importante que a velocidade sistólica máxima seja superior a 60 cm/s para se ter certeza de que a artéria uterina, e não a artéria arqueada, esteja sendo examinada.
Quando três formas semelhantes de onda são obtidas consecutivamente, o PI deve ser medido e o PI médio das artérias esquerda e direita calculado.
Índice de pulsatilidade (PI) = (Pico da velocidade sistólica máxima – velocidade diastólica mínima) / Velocidade média
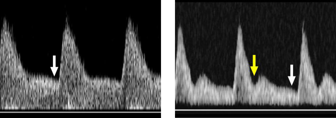
A forma da onda superior possui um bom fluxo diastólico final (seta branca). A forma da onda na parte inferior mostra um fluxo com alta resistência e com incisura diastólica precoce (seta amarela) e fluxo diastólico extremamente baixo (seta branca).
Em gestações normais, o UTPI está reduzido, refletindo a baixa impedância ao fluxo devido à transformação das artérias espiraladas, de vasos estreitos de alta resistência para canais dilatados de baixa resistência. Nas gestações com placentação deficiente e alto risco de desenvolver PE pré-termo, o UTPI é alto.
Em gestaç?es normais, o UTPI diminui com a idade gestacional e peso materno e é maior em mulheres de origem racial Afro-Caribenha do que em Caucasianas. Consequentemente, para o seu uso efetivo no rastreio, essas co-variáveis precisam ser levadas em consideração, padronizando os níveis do UTPI em valores normais de múltiplos da mediana (MoM).
Em gestaç?es que desenvolveram PE, o UTPI está aumentado e a diferença nos valores normais de MoM é maior em idades gestacionais precoces do que nas avançadas, quando o parto por conta da PE torna-se necessário. Consequentemente, o desempenho do rastreio é superior para PE pré-termo do que para PE a termo.
A inclinação das linhas de regressão dos MoM do UTPI em comparação com a idade gestacional ao parto, em gestações que desenvolvem PE, é maior conforme a idade gestacional. No entanto, as linhas de regressão cruzam 0 log MoM ao redor de 40 semanas e este marcador mostra pouca ou nenhuma força discriminatória para PE a termo. Consequentemente, o desempenho do rastreio melhora com o avanço da gravidez, mas o mesmo, é pior, independentemente da idade gestacional para PE pré-termo.
- Tayyar A, Guerra L, Wright A, Wright D, Nicolaides KH. Uterine artery pulsatility index in the three trimesters of pregnancy: effects of maternal characteristics and medical history. Ultrasound Obstet Gynecol 2015; 45: 689-97.
- O’Gorman N, Tampakoudis G, Wright A, Wright D, Nicolaides KH. Uterine artery pulsatility index at 12, 22, 32 and 36 weeks’ gestation in screening for pre-eclampsia. Ultrasound Obstet Gynecol 2016; 47: 565-72.
Fator de crescimento placentário (PLGF)
O PLGF é sintetizado pela placenta e possui potentes funções angiogênicas. Nas gestações que desenvolvem PE, o PLGF sérico é menor do que nas gestações normais; e esta diminuição é pressupostamente uma consequência da hipóxia placentária.
Na gravidez normal, o nível de PLGF sérico depende de fatores maternos e do aparelho utilizado para sua dosagem. Consequentemente, para o seu uso efetivo no rastreio, essas co-variáveis precisam ser levadas em consideração, padronizando os níveis de PLGF em valores de MoM.
Nas gestações que desenvolvem PE, o PLGF sérico está diminuído e a diferença nos valores normais de MoM é maior em idades gestacionais precoces do que nas avançadas, quando o parto por conta da PE torna-se necessário. Consequentemente, o desempenho do rastreio é superior para PE pré-termo do que para PE a termo.
A inclinação das linhas de regressão dos MoM do PLGF em comparação com a idade gestacional ao parto, em gestações que desenvolvem PE, é maior conforme a idade gestacional. Consequentemente, o desempenho do rastreio melhora com o avanço da gravidez.
- Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, Schisterman EF, Thadhani R, Sachs BP, Epstein FH, Sibai BM, Sukhatme VP, Karumanchi SA. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med 2004; 350: 672-83.
- Tsiakkas A, Duvdevani N, Wright A, Wright D, Nicolaides KH. Serum placental growth factor in the three trimesters of pregnancy: effects of maternal characteristics and medical history. Ultrasound Obstet Gynecol 2015; 45: 591-8.
- Tsiakkas A, Cazacu R, Wright A, Wright D, Nicolaides KH. Maternal serum placental growth factor at 12, 22, 32 and 36 weeks’ gestation in screening for pre-eclampsia. Ultrasound Obstet Gynecol 2016; 47: 472-7.
Serum soluble fms-like tyrosine kinase-1 (sFLT-1)
A sFLT-1 é um fator anti-angiogênico que supostamente exerce papel chave na patogênese da PE. A sFLT-1 exógena administrada a ratas grávidas induz hipertensão, proteinúria e endoteliose glomerular.
Na gravidez normal, o nível de sFLT-1 sérico depende de fatores maternos e do aparelho utilizado para sua dosagem. Consequentemente, para o seu uso efetivo no rastreio, essas co-variáveis precisam ser levadas em consideração, padronizando os níveis de sFLT-1 em valores de MoM.
Nas gestações com PE estabelecida, a sFLT-1 sérica está aumentada e esse aumento precede o desenvolvimento da doença em cerca de cinco semanas. Entre 11-13 semanas, a taxa de predição de PE obtida por fatores maternos não é maior quando somada a dosagem sérica da sFLT-1. A dosagem às 22 semanas é útil na predição de PE antes de 32 semanas (banda azul), a dosagem nas 32 semanas é útil na predição de PE antes de 37 semanas (banda azul) e a medição nas 36 semanas é útil na predição de PE após 37 semanas (banda azul).
- Maynard SE, Min JY, Merchan J, Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 2003; 111: 649-58.
- Tsiakkas A, Mendez O, Wright A, Wright D, Nicolaides KH. Maternal serum soluble fms-like tyrosine kinase-1 at 12, 22, 32 and 36 weeks’ gestation in screening for pre-eclampsia. Ultrasound Obstet Gynecol 2016; 47: 478-83
- Tsiakkas A, Duvdevani N, Wright A, Wright D, Nicolaides KH. Serum soluble fms-like tyrosine kinase-1 in the three trimesters of pregnancy: effects of maternal characteristics and medical history. Ultrasound Obstet Gynecol 2015; 45: 584-90.
Pregnancy associated plasma protein-A (PAPP-A)
A PAPP-A é produzida pela placenta e supostamente exerce um papel importante no crescimento e no desenvolvimento da placenta. Os níveis séricos maternos de PAPP-A no primeiro trimestre da gravidez diminuem em gestações com trissomias fetais do 21, 18 e 13.
Nas gestações que desenvolvem PE, em comparação com as gestações normais, a PAPP-A sérica materna está diminuída durante o primeiro trimestre, não significativamente diferente no segundo trimestre e aumentada no início do terceiro trimestre.
A diferença nos valores normais de MoM é maior em idades gestacionais precoces do que nas avançadas, quando o parto por conta da PE torna-se necessário; consequentemente, o desempenho do rastreio é superior para PE pré-termo do que para PE a termo.
Entre 11-13 semanas de gestação, a PAPP-A não melhora o desempenho do teste combinado com fatores maternos, UTPI, PAM e PLGF sérico. O mesmo, a não melhora do desempenho, é observado entre 30-34 semanas, com a associação da PAPP-A a fatores maternos, UTPI, PAM, PLGF e sFLT-1.
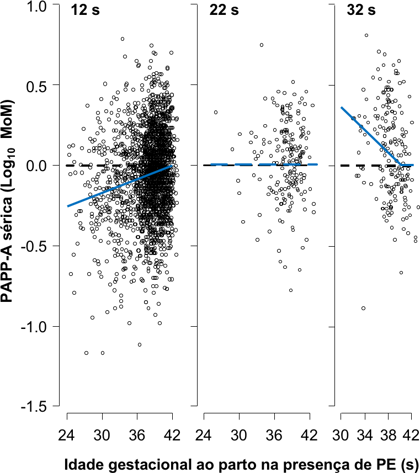
Viabilidade do rastreio combinado
Registro da história materna e a medida da pressão arterial são universalmente realizados como parte da rotina pré-natal.
A medida da PAM exige a aderência a um protocolo, mas pode ser realizada rapidamente e com o uso de equipamentos baratos, por assistentes de saúde após um treinamento mínimo.
A medida do UTPI exige treinamento específico de ultrassonografistas e garantia de qualidade de seus resultados. No entanto, este teste pode ser realizado dentro de alguns minutos pelos mesmos profissionais e máquinas como parte da ultrassonografia de rotina da gravidez.
A dosagem do PLGF sérico e da sFLT-1, apesar de acarretar um aumento inevitável no custo, pode ser realizada nos mesmos aparelhos usados para a leitura do ß-hCG e PAPP-A, amplamente utilizados no rastreio da síndrome de Down.
Em última análise, a escolha do teste para rastreio dependerá não apenas da performance, mas também da viabilidade de implementação e das considerações econômicas das políticas de saúde.
Desempenho do rastreio
Rastreio entre 11-13 semanas
O objetivo do rastreio de PE entre 11-13 semanas de gestação é identificar os casos que se beneficiariam do uso profilático de aspirina, reduzindo o risco de PE pré-termo em mais de 60%.
O rastreio combinado, que possui taxa de rastreio positivo de 10%, associando-se fatores maternos, PAM, UTPI e PLGF prevê cerca de 90% de PE precoce (<34 semanas), 75% de PE pré-termo (<37 semanas) e 45% de PE a termo (?37 semanas).
- A inclusão de PAPP-A e sFLT-1 não melhora o desempenho do rastreio.
A abordagem tradicional para identificar mulheres com alto risco de desenvolver PE e que poderiam se beneficiar da aspirina é baseada em fatores maternos. No Reino Unido, o Instituto Nacional de Saúde e Excelência Clínica (NICE) recomenda que a identificação do grupo de alto risco seja baseada em 10 fatores presentes nas características maternas e histórico médico; este método identifica cerca de apenas 40% dos casos de PE pré-termo e 35% de PE a termo. O Colégio Americano de Ginecologistas e Obstetras (ACOG) recomenda o uso de aspirina em mulheres com antecedente de PE em ?2 gestações ou com antecedente de PE e parto <34 semanas; este método identifica 5% dos casos de PE pré-termo e 2% de PE a termo.
- O’Gorman N, Wright D, Syngelaki A, Akolekar R, Wright A, Poon LC, Nicolaides KH. Competing risks model in screening for preeclampsia by maternal factors and biomarkers at 11-13 weeks’ gestation. Am J Obstet Gynecol 2016;214:103.e1-103.e12.
- National Collaborating Centre for Women’s and Children’s Health (UK). Hypertension in pregnancy: the management of hypertensive disorders during pregnancy. London: RCOG Press, 2010.
- Hypertension in pregnancy: report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet Gynecol 2013; 122: 1122-31.
- O’ Gorman N, Wright D, Poon LC, Rolnik DL, Syngelaki A, de Alvarado M, Carbone IF, Dutemeyer V, Fiolna M, Frick A, Karagiotis N, Mastrodima S, de Paco Matallana C, Papaioannou G, Pazos A, Plasencia W, Nicolaides KH. Multicenter screening for preeclampsia by maternal factors and biomarkers at 11-13 weeks’ gestation: comparison to NICE guidelines and ACOG recommendations. Ultrasound Obstet Gynecol 2017; 49: 756-60.
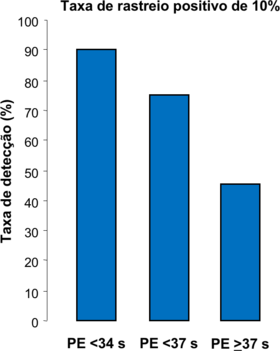
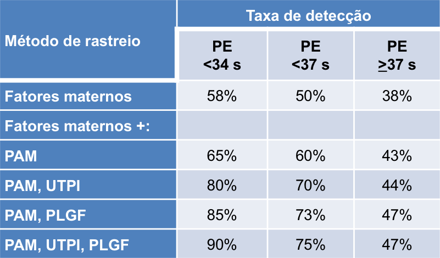
Rastreio entre 20-24 semanas
O objetivo do rastreio de PE entre 20-24 semanas de gestação é estimar o risco específico da paciente em desenvolver PE e, com base neste risco, definir o manejo subsequente da gravidez, incluindo o intervalo e o conteúdo das consultas seguintes. Isso poderia potencialmente minimizar eventos perinatais adversos naquelas que desenvolvem PE por meio do planejamento do momento e local do parto.
Nas gestações que desenvolvem PE, no segundo trimestre, os valores da PAM, UTPI e sFLT-1 estão aumentados e o PLGF diminuído. Para todos os biomarcadores, o desvio da normalidade é inversamente proporcional à idade gestacional em que o parto torna-se necessário por indicações maternas e / ou fetais e, portanto, o desempenho do rastreio é melhor para PE pré-termo do que para PE a termo.
O rastreio combinado por fatores maternos, PAM, UTPI, PLGF e sFLT-1 prevê todos os casos de PE precoce (<34 semanas), com taxa de rastreio positivo de 10%. A sFLT-1 sérica melhora o desempenho do rastreio para PE precoce, mas não para PE ?34 semanas. A associação de fatores maternos, PAM, UTPI e PLGF prevê 85% de PE pré-termo (<37 semanas) e 45% de PE a termo (?37 semanas).
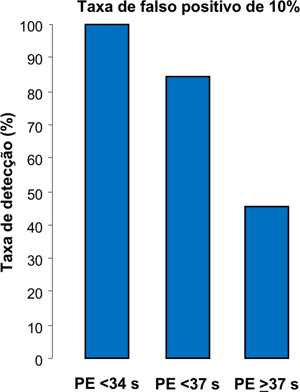
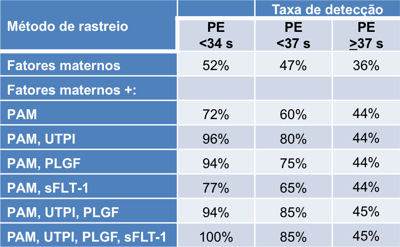
astreio entre 30-34 semanas
O objetivo do rastreio de PE entre 30-34 semanas de gestação é estimar o risco específico da paciente em desenvolver PE e, com base neste risco, definir o manejo subsequente da gravidez, incluindo o intervalo e o conteúdo das consultas seguintes. Isso poderia potencialmente minimizar eventos perinatais adversos naquelas que desenvolvem PE por meio do planejamento do momento e local do parto.
Nas gestações que desenvolvem PE, os valores da PAM, UTPI e sFLT-1 estão aumentados e o PLGF diminuído. Para todos os biomarcadores, o desvio da normalidade é inversamente proporcional à idade gestacional em que o parto torna-se necessário por indicações maternas e / ou fetais e, portanto, o desempenho do rastreio é melhor para PE pré-termo do que para PE a termo.
O rastreio combinado por fatores maternos, PAM, UTPI, PLGF e sFLT-1, com taxa de rastreio positivo de 5%, prevê quase todos os casos de PE pré-termo (<37 semanas), mas apenas 55% de PE a termo (?37 semanas).
- Os biomarcadores importantes nessa idade gestacional são PAM, PLGF e sFLT-1, sendo que a inclusão do UTPI adiciona pouco benefício.
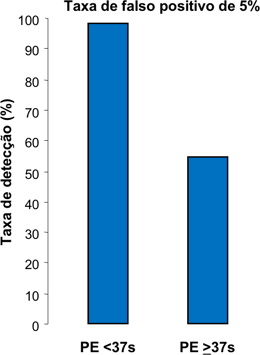
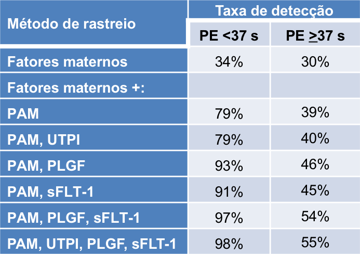
Rastreio entre 35-37 semanas
O objetivo do rastreio de PE entre 35-37 semanas de gestação é estimar o risco específico da paciente em desenvolver PE e, com base neste risco, definir o manejo subsequente da gravidez, incluindo o intervalo e o conteúdo das consultas seguintes. Isso poderia potencialmente minimizar eventos perinatais adversos naquelas que desenvolvem PE por meio do planejamento do momento e local do parto.
Nas gestações que desenvolvem PE, os valores da PAM, UTPI e sFLT-1 estão aumentados e o PLGF diminuído.
O rastreio combinado por fatores maternos, PAM, UTPI, PLGF e sFLT-1, taxa de rastreio positivo de 10%, prevê cerca de 85% dos casos de PE a termo (?37 semanas).
- Os biomarcadores importantes nessa idade gestacional são PAM, PLGF e sFLT-1, sendo que a inclusão do UTPI adiciona pouco benefício.
A taxa de detecção de PE a termo, com taxa de rastreio positivo de 10%, aumentou de mais ou menos 45% com o rastreio combinado entre 12 e 22 semanas para cerca de 65% com o rastreio entre 32 semanas e para 85% com o rastreio com 36 semanas.
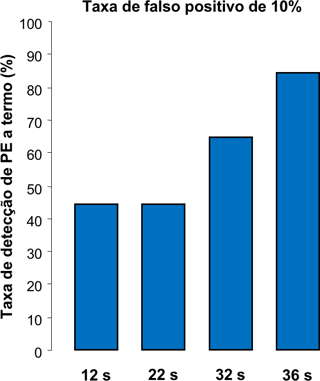
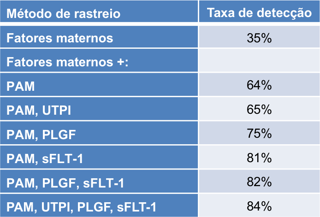
Estratificação do manejo da gravidez
11-13 semanas de gestação
O rastreio de toda a população pode ser realizado por uma combinação de fatores maternos, PAM, UTPI e PLGF e, com base nos riscos em desenvolver PE, as gestantes são estratificadas em duas vias possíveis:
- Grupo de alto risco (risco de PE <37 semanas de ?1 em 100). Este grupo constitui cerca de 10% do total e contém 75% daquelas que desenvolvem PE antes de 37 semanas. O tratamento com aspirina (150 mg/dia) tomada à noite entre 12 e 36 semanas reduz o risco de PE pré-termo em cerca de 60%. As pacientes devem ser encorajadas a tomar o medicamento regularmente, porque se a adesão for ?90%, a redução do risco pode ser de 75%.
- Grupo de baixo risco (risco de PE <37 semanas de <1 em 100). Este grupo, que constitui 90% do total, pode ser tranquilizado, uma vez que, o desenvolvimento de PE antes de 37 semanas é improvável, mas uma nova avaliação do risco às 22 semanas de gestação é necessária.
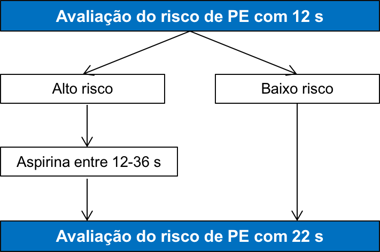
20-24 semanas de gestação
O rastreio de toda a população pode ser realizado por uma combinação de fatores maternos, PAM, UTPI e PLGF e, com base nos riscos em desenvolver PE, as gestantes são estratificadas em diferentes vias de manejo, incluindo reavaliação do risco e monitoramento intensivo em vários períodos da gestação. O objetivo seria maximizar, com o menor custo possível, a detecção de PE, tendo em vista a proporção da população que necessita uma avaliação adicional e aqueles que necessitam monitoramento intensivo.
- Grupo de alto risco (risco de PE <32 semanas de ?1 em 100). Este grupo representa cerca de 1% do total e contém 95% daquelas que desenvolvem PE antes de 32 semanas. Estas gestantes exigem monitoramento regular com medida da pressão arterial e análise de urina entre 24-31 semanas. Monitoramento intensivo adicional pode ser necessário para aquelas que apresentam à ultrassonografia de 22 semanas sinais de restrição de crescimento fetal intrauterino e aquelas que desenvolvem hipertensão.
- Grupo de baixo risco (risco de PE <36 semanas de <1 em 300). Este grupo, que representa mais de 80% do total, pode ser assegurado que o desenvolvimento de PE antes de 36 semanas é improvável, mas uma reavaliação do risco com 36 semanas é necessária.
- Grupo de risco intermediário (risco entre os grupos de alto e baixo risco acima). Este grupo, juntamente com as pacientes do grupo de alto risco que permanecem grávidas após 32 semanas, constitui <20% do total e contém >90% das gestações que desenvolvem PE entre 32-35 semanas. Estas gestantes necessitam de um novo cálculo de risco com 32 semanas de gravidez.
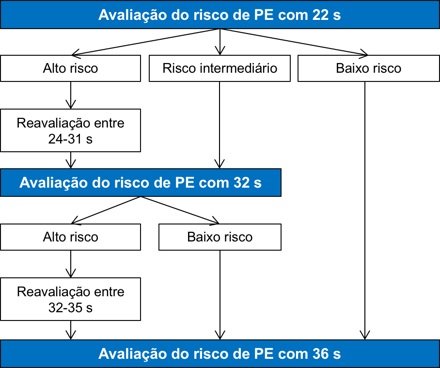
36 semanas de gestação
O rastreio de toda a população pode ser realizado por uma combinação de fatores maternos, PAM, UTPI e PLGF e, com base nos riscos em desenvolver PE, as gestantes são estratificadas em diferentes vias de manejo:
- Grupo de alto risco (risco de PE <40 semanas de ?1 em 100). Este grupo representa cerca de 20% da população total e contém 90% daquelas que desenvolvem PE antes de 40 semanas. Estas gestantes exigem monitoramento regular com medida da pressão arterial e análise de urina entre 36-39 semanas. Monitoramento intensivo adicional pode ser necessário para aquelas que apresentam à ultrassonografia de 36 semanas sinais de restrição de crescimento fetal intrauterino e aquelas que desenvolvem hipertensão.
- Grupo de baixo risco (risco de PE <42 semanas de <1 em 200). Este grupo, que representa cerca de 40% do total, possui 99,9% de chance de não desenvolver PE e, na ausência de qualquer outra indicação obstétrica, pode ser orientado a aguardar o trabalho de parto espontâneo.
- Grupo de risco intermediário (risco entre os grupos de alto e baixo risco acima). Este grupo, juntamente com as pacientes do grupo de alto risco que atingiram 40 semanas de gestação e ainda permanecem grávidas, constitui cerca de 60% do total e contém quase todos os casos de PE após 40 semanas. Estas gestantes requerem uma nova avaliação com 40 semanas para o planejamento do parto.
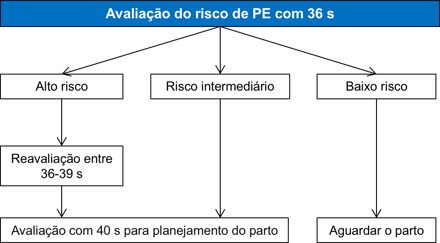
Agradecimentos
Diretor do curso: Kypros Nicolaides
Colaboradores: Despoina-Antigoni Nikolaidi, Luciana Francine Bocchi De Stefani
Programação: Dimitris Vichos
Table of Contents
Restrição de crescimento fetal [+]
Isoimunização de células vermelhas [+]
Taquicardia supraventricular [+]
Acompanhamento Antenatal
Visão Geral
O acompanhamento antenatal tem por objetivo a prevenção de morte e deficiências decorrentes do período perinatal. É planejado melhor quando os fatores de risco maternos e as características da condição fetal são levadas em consideração.
O acompanhamento deve ser específico à cada doença e requer:
-
- Reconhecimento de fatores de risco maternos específicos.
- Compreensão da evolução clínica da doença materna.
- Compreensão da evolução clínica da doença fetal
- Avaliação física do feto
- Execução e interpretação precisas dos testes de acompanhamento
Em cada visita da paciente , as principais decisões são:
-
- Existe indicação para o parto?
- Existe indicação para corticosteróides?
- Quando devo ver a paciente novamente ?
Professor Ahmet Baschat
Diretor de terapia fetal ,
Universidade John Hopkins
Visão Geral
O acompanhamento antenatal deve conter as seguintes informações:
- História materna e fatores de risco
- Avaliação fetal
- Anatomia
- Tamanho e proporção
- Velocidade de crescimento
- Volume de líquido amniótico
- Variáveis biofísicas
- Parâmetros de frequência cardíaca
- Parâmetros cardiovasculares
Ênfase em determinados testes é baseada em condições específicas
Fatores de risco materno
Conhecer os fatores de risco maternos é essencial pois determina quais testes devem ser realizados e com que frequência:
- Uma história e exame físico minuciosos devem fazer parte da avaliação inicial da paciente
- Exames de laboratório podem ser indicados para esclarecer diagnósticos e prognósticos
As pacientes devem ser classificadas de acordo com os problemas em gestações anteriores ou na atual e de acordo com doenças crônicas:
- Gestação atual: hipertensão, pré-eclampsia, diabetes gestacional.
- Gestações anteriores: pré-eclampsia, abortos ou natimortos, descolamento de placenta
- Doenças crônicas: hipertensão, diabetes, lúpus, trombofilias
Anatomia fetal
Defeitos cromossômicos, síndromes fetais e infecções virais simulam condições fetais potencialmente tratáveis. É, portanto, essencial realizar uma ultrassonografia detalhada para:
Características de aneuploidias
-
- Múltiplas anomalias
- Múltiplos marcadores
- Crescimento alterado
Características de síndromes fetais
-
- Combinações de anomalias fetais reconhecidas
Infecções virais
-
- Hiperecogenicidade em diferentes orgãos
- Acúmulo de flúido em cavidades no corpo fetal
- Crescimento alterado
Estes diagnósticos diferenciais devem ser considerados à cada visita
Tamanho e proporção fetal
O tamanho fetal é avaliado medindo-se a cabeça, circunferência abdominal e membros. A avaliação do tamanho requer tabelas de referência e conhecimento da idade gestacional
- Tamanho da cabeça: diâmetro biparietal (DBP), circunferência cefálica (CC), diâmetro cerebelar (DC)
- Tamanho do corpo: circunferência abdominal (CA)
- Tamanho dos membros : Comprimento do fêmur(CF), Comprimento do úmero (UC)
- Peso fetal estimado: calculado por diferentes associações entre as medidas da cabeça, corpo e ossos longos fetais
O crescimento fetal anormal é subdividido em assimétrico e simétrico baseado na relação cabeça-circunferência abdominal (CC/CA, DC/CA), relação cabeça-fêmur (CC/CF) e relação circunferência abdominal – fêmur (CA/CF):
Tipo Assimétrico:
-
- Restrição de crescimento fetal devido a implantação inadequada da placenta ou triploidia (relação CC/CA aumentada)
- Displasias esqueléticas (Relação CC/CF e CA/CF aumentadas)
- Microcefalia(relação CC/CA e CC/CF diminuídos), macrossomia fetal relacionada a diabetes materna(Relação CC/CA e CC/CF diminuídas)
Tipo Simétrico (CC/CA normal):
-
- Restrição de crescimento fetal grave devido a implantação inadquada da placenta
- Aneuploidias, síndromes genéticas, infecções congênitas
Crescimento anormal
O crescimento é dinâmico e medidas seriadas em intervalos de 2-3 semanas são necessárias para definir normalidade e anormalidade:
- Crescimento normal é indicado por medidas da cabeça, circunferência abdominal e membros dentro dos limites normais para a idade gestacional.
- Crescimento anormal é indicado por medidas da cabeça e/ou circunferência abdominal e/ou membros abaixo dos limites normais para a idade gestacional
Padrões de crescimento anormal:
- Crescimento anormal da cabeça sugere microcefalia. Possíveis causas incluem holoprosencefalia, aneuploidia ou infecções virais
- Circunferência abdominal é a melhor medida isolada do estado nutricional fetal. Uma medida abdominal pequena sugere restrição de crescimento fetal, podendo ser simétrica se a ciscunferência cefálica também for pequena, ou assimétrica se a circunferência cefálica for normal.
- Membros curtos com as circunferências cefálica e abdominal dentro dos limites normais sugere a presença de displasias esqueléticas.
Líquido amniótico
O líquido amniótico é constituído de urina fetal , porém, nas primeiras 16 semanas, fontes adicionais de flúido incluem a placenta, membranas amnióticas, cordão umbilical e a pele fetal. O líquido amniótico é removido pela deglutição fetal.
Avaliação do líquido amniótico é geralmente subjetiva. Quantitativamente, é definido tanto pela medida da distância vertical do maior bolsão de líquido sem partes fetais , ou ainda pelo índice de líquido amniótico, o qual é a soma das medidas verticais dos maiores bolsões de líquido de cada quadrante do útero.
Oligodramnia. As principais causas são: ruptura das membranas, insuficiência uteroplacentária, anomalias do trato urinário e o feto doador na síndrome de transfusão feto-fetal.
- O diagnóstico de oligodrâmnio é feito, geralmente, subjetivamente pela ausência da “ janela acústica” ,normalmente fornecida pelo líquido amniótico, e pela posição fletida do feto com ausência dos seus movimentos. Os critérios qualitativos são: o maior bolsão vertical medindo menos de 2cm ou o índice de líquido amniótico abaixo do percentil 5 do intervalo normal
Polidramnia. Existem essencialmente duas causas maiores de polidramnia:
- Diminuição da deglutição fetal, devido a defeitos crânio-espinhais (como anencefalia), tumores faciais, obstruções gastrointestinais, compressão pulmonar e outras anomalias torácicas, e acinesia fetal (devido ao comprometimento neuromuscular da deglutição fetal )
- Aumento da micção fetal, devido a diabetes mellitus materna, circulação fetal hiperdinâmica secundária a anemia fetal , tumores placentários e fetais ou malformações artério-venosas, e ainda no feto recipiente na síndrome de transfusão feto-fetal.
A medida vertical do único maior bolsão de líquido amniótico é usada para classificar polidramnia em leve (8-11 cm), moderada (12-15 cm) e grave (16 cm ou mais)
Dopplerfluxometria
Uma abordagem padronizada para a Dopplerfluxometria de todos os vasos é essencial para alcançar resultados reprodutíveis e confiáveis:
- Zoom na área de interesse
- Aplicar o color Doppler
- Diminua a área da caixa de cor
- Ajuste a escala de velocidade
- Aplicar o Doppler pulsátil
- Ajuste o volume de amostra para cobrir o vaso
- Ajuste a escala de velocidade
- Ajuste o filtro
- Adquira ao menos três ondas de fluxo uniformes, sem movimentos fetais ou movimentos respiratórios fetais.
Dopplerfluxometria
Na avaliação da impedância ao fluxo:
- É importante obter um traçado contínuo da onda de fluxo , do começo até o início da próxima. Nos vasos arteriais, traçados automáticos podem ser utilizados, porém nas veias a onda trifásica não é devidamente avaliada e sendo assim o traçado manual deve ser utilizado.
- Tanto em artérias quanto em veias a medida do índice de pulsatilidade (IP) é recomendado e curvas de normalidades devem ser utilizadas para interpretar os resultados.
Na avaliação de uma circulação hiperdinâmica, como na anemia fetal, o pico de velocidade sistólica (PVS), o qual é o ponto mais alto da onda ao Doppler, deve ser considerado.
Calculadora de Dopplerfluxometria
Você pode digitar as medidas Doppler da sua paciente e vê-las graficamente em uma curva de normalidade dos índices de pulsatilidade das artérias uterinas, umbilical, cerebral média e ducto venoso; e pico de velocidade sistólica da artéria cerebral média.
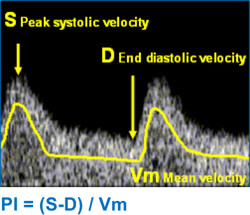
Doppler arterial
As relações de velocidade sistólica e diastólica e as características da onda de fluxo dependem de:
- Pressão de entrada
- Resistência vascular
Resistência vascular pode ser alterada por:
- Mudanças no tônus vascular
- Autoregulação das artérias cerebral média, renal, hepática, adrenal, coronárias e esplênicas
- Vasoconstrição do ducto arterioso após a administração de indometacina
- Mudanças na estrutura vascular das artérias uterinas e umbilicais na implantação inadequada da placenta
Doppler venoso
Clinicamente , os vasos mais estudados são o ducto venoso, veia cava inferior e veia umbilical
O Doppler venoso fornece informações sobre função cardíaca de ejeção
- Complacência
- Relaxamento
- Contratilidade
- Pós carga
Todos os vasos têm o mesmo formato de onda
- Pico sistólico
- Pico Diastólico
- Sístole atrial
Doppler úteroplacentário
A placenta é um órgão de troca de gás, flúido e nutrientes com dois compartimentos:
- O compartimento materno é avaliado pelo Doppler das artérias uterinas
- O compartimento fetal é avaliado pelo Doppler das artérias umbilicais
Maturação fisiológica da vascularização com o avançar da gravidez é refletido em:
- Artérias uterinas: aumento do fluxo diastólico com a perda da incisura no início da diástole.
- Artérias umbilicais: Surgimento de velocidade de fluxo no fim da diástole
- Redução do índice de pulsatilidade em ambos os vasos
Na placentação anormal existe:
- Invasão trofoblástica deficiênte nas artérias espirais maternas, resultando em um índice de pulsatilidade alto e a persistência da incisura na artéria uterina
- A árvore vascular anormal nas vilosidades resulta em onda de fluxo anormal nas artérias umbilicais, a qual depende do grau de comprometimento da vascularização:
- 30% de comprometimento resulta em índice de pulsatilidade alto
- 50% de comprometimento resulta em ausência de fluxo na diástole ( diástole zero)
- 70% de comprometimento resulta em revesão da onda de fluxo na diástole (diástole reversa)
O risco de hipoxemia/ acidemia é proporcional à diminuição do fluxo diastólico na artéria umbilical

A circulação fetal
Circulação paralela com fluxo preferencial através do:
- Ducto venoso, o qual determina fluxo de nutrientes entre fígado e coração
- Forâme oval, o qual determina o equilíbrio entre coração direito e esquerdo
- Ducto arterioso, o qual determina o equilíbrio de suprimento de nutrientes para o cérebro versus o corpo
Alteração no ducto venoso afeta o equilíbrio de toda a circulação subsequente
Mudanças na resistência do fluxo arterial afetam o equilíbrio entre o coração, cérebro e corpo
Veia umbilical
A veia umbilical pode ser examinada tanto no abdome fetal quanto no cordão umbilical
O fluxo é constante a partir de 12 semanas em 90% dos fetos
Pulsatilidade é observada em 10% dos fetos e pode ser:
- Monofásico
- Bifásico
- Trifásico
Fluxos monofásicos são relevantes se as veias centrais estão anormais
Fluxos multifásicos indicam pressão venosa aumentada anormalmente
Aplicação clínica do Doppler de veia umbilical: restrição de crescimento fetal, síndrome de transfusão feto-fetal e hidropsia fetal
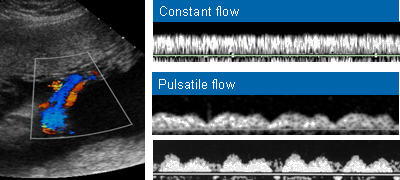
Ducto venoso
O ducto venoso é o principal shunt regulador do fluxo de nutrientes para o coração e fígado
Esse vaso pode ser visto num corte sagital ou transverso do abdome
- Em fetos normais a onda-a é positiva a partir do primeiro trimestre e o índice de pulsatilidade venoso diminúi com o avançar da gestação
- Índice de pulsatilidade venoso aumentado e onda-a ausente ou reversa são observados em fetos com aneuploidias, defeitos cardíacos, restrição de crescimento fetal e tanto do recipiente quanto no doador na síndrome de transfusão feto-fetal.
Doppler do ducto venoso
Artéria cerebral média
A artéria cerebral média (ACM) é um ramo do polígono de Willis, na base do crânio
Um corte transverso da cabeça fetal deve ser obtido e o mapeamento com color Doppler deve ser usado para identificar o polígono de Willis e a ACM. O volume de amostra do Doppler pulsátil é então colocado no terço proximal da ACM. O ângulo de insonação deve ser zero graus. Deve se tomar cuidado para evitar pressão desnecessária na cabeça fetal pois isso aumenta tanto o pico de velocidade sistólico (PVS) quanto o índice de pulsatilidade (PI)
- Em gestações normais o PVS aumenta e o IP diminúi com o avançar da gestação
- Redução do IP é observado em hipoxemia fetal e hipertensão fetal
- Aumento do PVS é observado em anemia fetal e em hipercapnia (aumento do pCO2)
Doppler da artéria cerebral média
Comportamento fetal
O comportamento fetal se desenvolve sequencialmente como o avançar da gestação
Primeiro trimestre: Movimentos corporais e respiratórios espontâneos e isolados, sem relação um com o outro e sem relação com estímulos
Segundo trimestre: Desenvolvimento de comportamento responsivo , ou seja, atividade em resposta a estímulos. Por exemplo, aceleração da frequência cardíaca em resposta à movimentos fetais, aumento dos movimentos respiratórios em resposta à hiperglicemia materno-fetal, movimentos reativos tipo “susto” em resposta a estímulo vibro-acústico
Terceiro trimestre: Desenvolvimento de estados comportamentais regulados pelo cérebro fetal. Os estados são distintos entre sono e vigilância e a sua duração aumenta com o avançar da gestação. O sono é caracterizado pela ausência de movimento e redução da variabilidade da frequência cardíaca fetal (FCF). Em contraste, a vigilância é caracterizada pela movimentação fetal ativa , reativa e padrão de FCF reativa.
Na avaliação individual de cada feto é importante manter em mente que os centros de regulação no cérebro dependem de, primeiramente, maturação e portanto da idade gestacional, e secundariamente, na oxigenação adequada. No acompanhamento fetal a ausência de parâmetros individuais (movimentos fetais, respiratórios, reatividade de FCF) é mais provável que seja consequência de um período de descanso. Em contraste, a hipóxia fetal é caracterizada pela ausência concomitante de múltiplos parâmetros
Tônus fetal, movimentos corporais e respiratórios
Movimentos corporais fetais: caracterizado por movimentos ativos da cabeça, tronco e/ou membros
Tônus fetal: Refere-se a habilidade do feto em permanecer na posição fletida e em demonstrar movimentos das mãos e pés. Ausência de tônus é diagnosticada se há ausência de qualquer atividade e as mãos permanem abertas
Movimentos respiratórios fetais: Refere-se a períodos de movimentos de expansão do tórax e/ou diafragma. Soluços também são considerados como movimentos respiratórios
Ausência de tônus fetal, movimentos respiratórios e corporais podem ser explicados por:
- Hipoxemia fetal e acidemia
- Repouso fetal
- Medicações interferindo com a trasmissão neuromuscular, como magnésio, diazepam e nifedipina
- Anomalias neuromusculares e do cérebro
Além disso, movimentos respiratórios podem estar ausentes durante o período de jejum materno
Ausência de tônus fetal e movimentos corporais são interpretados no contexto de um perfil biofísico completo. A ausência de movimentos respiratórios fetais deve ser reavaliada após a gestante comer.
Frequência cardíaca fetal
O padrão da frequência cardíaca fetal (FCF) proporciona um registro da regulação autonômica da atividade cardíaca intrínseca e sua modulação por centros reguladores:
- Centro vasomotor
- Sistema de ativação reticular
- Sistema nervoso autônomo
O padrão de FCF pode ser analisada :
- Visualmente descrevendo a linha de base, reatividade, variabilidade e periodicidade de mudanças
- Automaticamente por medidas computadorizadas da variação de curto prazo (ms)
Interpretação dos padrões de FCF:
- Reatividade, praticamente exclui hipoxemia. As causas de não reatividade são idade gestacional precoce, estado comportamental de repouso, hypoxexemia ou acidemia e medicações
- Desacelerações variáveis são causadas por compressão do cordão umbilical
- Desacelerações tardias são sugestivas de hipoxemia fetal com uma queda maior que 8 torr na paCO2
- Redução na variabilidade de curto prazo para menos de 3,5 ms é sugestivo de hipoxemia fetal e desenvolvimento anormal do cérebro
Pontuação no perfil biofísico
Um sistema de pontuação composto por cinco variáveis onde cada uma vale 2 pontos pela presença e zero pela ausência :
Tônus: ao menos um episódio de extensão ativa dos membros, tronco ou mãos com retorno a posição de flexão
Movimentos: ao menos 3 movimentos discretos de corpo\membros
Respiração: ao menos um episódio de pelo menos 30 segundos de duração
Líquido amniótico: maior bolsão >2cm
Frequência cardíaca: ao menos 2 acelerações de:
-
- 10 batimentos para cima da linha de base, com duração de 10 segundos (24-28 semanas)
- 15 batimentos para cima da linha de base, com duração de 15 segundos (28-34 semanas)
- 20 batimentos para cima da linha de base, com duração de 20 segundos (após 34 semanas)
Conduta na gravidez. Existe uma boa correlação entre a pontuação total e a taxa de mortalidade perinatal na semana seguinte.
- Se a pontuação for de 0 a 2 é recomendado que o parto seja realizado imediatamente. Se a pontuação for de 4 a 6 com oligodrâmnio, o teste deve ser repetido imediatamente por uma hora e se não houver melhora é recomendado que o parto seja realizado imediatamente.
- Se o teste obtiver um resultado inconclusivo, um novo teste em 24 horas é recomendado
- Se o resultado for normal o teste é repetido, geralmente, em uma semana

Teste fetal integrado
Uma nova abordagem ao acompanhamento fetal é integrar as informações obtidas pela biometria, volume de líquido amniótico, perfil bioísico e Dopplerfluxometria das circulações fetais e placentárias. O motivo que torna a avalição integrada superior aos testes individuais é porque fetos comprometidos vão demostrar diferentes características dependendo da idade gestacional e a doença de base reponsável pelo comprometimento.
- Placentação anormal é manifestada com crescimento restrito assimétrico, perfil biofísico e índices de Doppler anormais
- Na acinesia fetal, a biometria é normal, o volume de líquido amniótico está aumentado, os índices de Doppler estão normais, porém os movimentos corporais e respiratórios fetais estão ausentes
- Na isoimunização de células vermelhas, o crescimento fetal, líquido amniótico e a impedância do fluxo na circulação placentária e fetal são normais e as principais características de anemia severa são o aumento do pico de velocidade sistólica na artéria cerebral média e redução da atividade fetal.
- Na diabetes mellitus materna sem vasculopatia e em diabetes gestacional, o comprometimento fetal é essencialmente a consequência da hiperglicemia manifestada como macrossomia fetal e polidramnia com perfil biofísico fetal e índices de Doppler normais. Em contraste, em diabetes com doença renal e vasculopatia as consequências fetais podem ser similares às observadas nos casos de placentação anormal.

Sumário
Testes fornecem informações específicas:
- Anatomia fetal ajuda no diagnóstico diferencial de restrição de crescimento fetal
- Crescimento fetal é um indicador do funcionamento da placentário
- Volume de líquido amniótico serve para avaliar as trocas placentárias
- Índice de pulsatilidade das artérias uterinas reflete o grau de invasão trofoblástica das artérias espiraladas
- Índice de pulsatilidade das artérias umbilicais reflete a área de troca vascular
- Índice de pulsatilidade da artéria cerebral média e o pico de velocidade sistólico refletem o pCO2 fetal, concentração de hemoglobina, oxigênio e hipertensão
- Doppler venoso reflete a função de ejeção cardíaca
- Variáveis dinâmicas indicam maturação, estado comportamental e oxigenação
- Padrão de frequência cardíaca fetal reflete oxigenação e desenvolvimento cerebral
Condições específicas requerem testes específicos
Restrição de crescimento fetal
Definição
Restrição de crescimento fetal (RCF) é definido pelo achado de crescimento abaixo do potencial genético esperado
O parâmetro isolado mais sensível e clinicamente factível no diagnóstico de RCF é a medida da circunferência abdominal (CA) abaixo do percentil 5 do limite de normalidade para a idade gestacional
Os diagnóstico diferenciais incluem :
Erro na datação: o feto é anatomicamente normal, as medidas são simetricamente pequenas, o líquido amniótico, atividade fetal e Doppler placentário são normais. Novo exame em duas semanas mostra uma velocidade de crescimento normal
Pequeno normal: o feto é anatomicamente normal, as medidas são simetricamente pequenas, o líquido amniótico, atividade fetal e Doppler placentário são normais. Novo exame em duas semanas mostra uma redução da velocidade de crescimento
Pequeno desnutrido: o feto é anatomicamente normal, as medidas são assimetricamente pequenas com o abdomen e o fêmur sendo mais afetados do que a cabeça; líquido amniótico e atividade fetal estão reduzidos, o índice de pulsatilidade da artéria uterina e/ou artérias umbilicais estão aumentados e na artéria cerebral média está reduzido. O padrão de frequência cardíaca fetal e/ou o fluxo no ducto venoso podem estar anormais. Novo exame em duas semanas pode demonstrar uma redução marcante da velocidade de crescimento e piora da condição fetal
Pequeno anormal: o feto pode apresentar anomalias sugestivas de aneuploidias, síndromes genéticas ou infecções congênitas. As medidas podem ser simetricamente pequenas ou pode haver redução assimétrica da cabeça ou membros. O volume de líquido amniótico e atividade fetal podem estar diminuídos, aumentados ou normais. O padrão de frequência cardíaca fetal e os índices de Doppler placentário e fetal podem estar normais ou alterados. Novo exame em duas semanas pode demonstrar uma redução na velocidade de crescimento
O diagnóstico diferencial e a progressão clínica fornecem o fundamento para o uso integrado das modalidades de monitoramento
Monitoramento
Intervalo de monitoramento: deve ser inversamente relacionado à gravidade do comprometimento fetal e à velocidade de deteriorização da condição clínica. Piora da condição fetal é sugerida por:
- Oligodramnia
- Artéria cerebral média: diminuição do índice de pulsatilidade (IP)
- Artéria umbilical: aumento do IP, fluxo diastólico ausente ou reverso
- Ducto venoso: aumento do IP, onda-a ausente ou reversa
Alterações que precedem óbito fetal: avaliações seriadas em gestações que resultaram em natimortos demonstraram diferenças importantes nas respostas nos últimos três dias que precedem o óbito, dependendo da idade gestacional:
- Em natimortos antes de 34 semanas há um aumento importante do IP nas artérias umbilicais e ducto venoso; diminuição do volume de líquido amniótico, tônus fetal e movimentos. A frequência de monitoramento deve aumentar no caso de piora do IP da artéria umbilical e/ou do ducto venoso ou diminuição do volume de líquido amniótico, podendo variar de uma vez por semana até diariamente
- Em natimortos após 34 semanas há uma redução do IP da artéria cerebral média, variação da frequência cardíaca fetal e volume de líquido amniótico. Deve se ter atenção especial com o IP da artéria cerebral média e o parto deve ser considerado se houver redução do IP, mesmo se os valores permanecerem acima do percentil 5
Quando indicar o parto
O objetivo do monitoramento antenatal é decidir o melhor momento para o parto, contrapondo o risco fetal versus o risco neonatal.
A chance de sobrevivência sem comprometimentos aumenta substancialmente com a idade gestacional em cerca de 2% por dia entre 24 e 28 semanas, e em cerca de 1% por dia até 32 semanas. Consequentemente, o limiar para o parto deve ser inversamente relacionado à idade gestacional
Após 34 semanas: índice de pulsatilidade (IP) aumentado nas artérias umbilicais ou IP aumentado no ducto venoso ou IP diminuído na artéria cerebral média ou índice de líquido amniótico abaixo do percentil 5.
31-33 semanas: fluxo diastólico ausente na artéria umbilical ou onda-a ausente no ducto venoso ou maior bolsão de líquido amniótico menor que 2 cm e ausência de movimentos
28-30 semanas: onda-a reversa no ducto venoso ou fluxo diastólico reverso na artéria umbilical e maior bolsão de líquido amniótico menor que 2 cm e ausência de movimentos
Menos que 28 semanas: onda-a reversa no ducto venoso e fluxo diastólico reverso na artéria umbilical e maior bolsão de líquido amniótico menor que 2 cm e ausência de movimentos
Gêmeos Monocoriônicos
Placentação anormal
Em gestações normais cada cotilédone é suprido por uma artéria umbilical, que carreia o sangue desoxigenado vindo do feto para a placenta, e por uma veia umbilical que transporta o sangue oxigenado da placenta para o feto.
Em gêmeos monocoriônicos, a maioria dos cotilédonos são normais (supridos por uma artéria e uma veia do mesmo feto) porém existem também comunicações vasculares anormais entre as circulações dos dois fetos:
- Alguns dos cotilédonos são divididos entre os dois fetos com um suprimento arterial de um dos gêmeos e a drenagem venosa do outro
- Existem também comunicações diretas artério-arteriais e veno-venosas entre os fetos.
- Fluxo sanguíneo não balanceado através dessas comunicações vasculares placentárias de um feto, doador, para outro, receptor, resulta em síndrome de transfusão feto-fetal (STFF).
Gêmeos Monocoriônicos
Complicações
Síndrome de transfusão feto-fetal (STFF): em cerca de 80% dos gêmeos monocoriônicos há equilíbrio na rede de fluxo sanguíneo através das conexões vasculares placentárias. No entanto, em cerca de 20% dos casos existe um desequilíbrio no fluxo de um feto (doador) para o outro (recipiente) resultando na STFF. Metade destes casos ( 10% do total) desenvolve STFF severa nas 16-24 semanas, com polidrâmnio no saco gestacional do recipiente poliúrico e oligodrâmnio no saco gestacional do doador anúrico. A doença foi dividida em quatro estágios (classificação de Quintero). No estágios 1 ( polidrâmnio / oligodrâmnio) e no estágio 2 (polidrâmnio / anidrâmnio com a bexiga permanecendo vazia no doador) não há comprometimento cardiovascular nos fetos, diferente do estágio 3 (fluxo diastólico ausente ou reverso nas artérias umbilicais do doador e / ou onda-a reversa ou ausente no ducto venoso do recipiente) e estágio 4 (recipiente com hidropsia)
Restrição de crescimento fetal seletiva (RCFs): outra complicação que acontece em cerca de 10% dos gêmeos monocoriônicos é o crescimento insuficiênte de um dos fetos (RCF seletiva) devido a divisão desigual da placenta entre os fetos. Neste caso um dos fetos cresce normalmente com atividade, índices de Doppler e líquido amniótico normais. O feto com RCF demonstra as características de uma insuficiência placentária, incluíndo crescimento assimétrico, relação cabeça-circunferência abdominal aumentada, atividade e líquido amniótico reduzidos e evidências de redistribuição na circulação fetal ao Doppler com índice de pulsatilidade (IP) aumentado na artéria umbilical e ducto venoso e IP diminuído na artéria cerebral média.
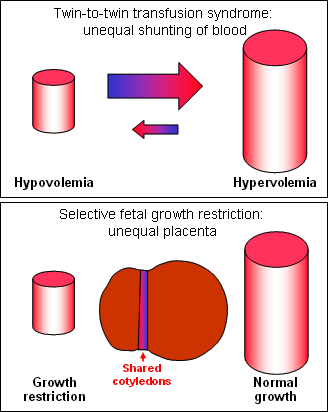
Monitoramento e terapia
Os riscos em gêmeos monocoriônicos (MC) são devidos, principalmente, às anastomoses vasculares e o compartilhamento da placenta entre os fetos e incluem:
- Síndrome de transfusão feto-fetal (STFF)
- Restrição de crescimento fetal (RCF)
- Morbidade neurológica ou morte em um ou ambos os fetos
Monitoramento em gêmeos MC deve integrar as seguintes informações:
- Dinâmica de crescimento e parâmetros biofísicos (especialmente em restrição de crescimento)
- Status de volume fetal (índice de líquido amniótico e enchimento de bexiga)
- Parâmetros vasculares (artéria umbilical, artéria cerebral média e Doppler venoso)
Terapia eficiente em gêmeos MC com STFF e RCF seletiva é realizada com separação das anastomoses vasculares placentárias por endoscopia e laser. Em ambas as condições essa separação previne os possíveis riscos de morte e sequelas em um dos fetos no caso do outro feto morrer. Adicionalmente, em caso de STFF a separação à laser interrompe a causa básica da doença e melhora o prognóstico. Terapia à laser provou ser mais efetiva do que o método anterior de drenagem amniótica seriada e é associada a 85% de sobrevivência de ao menos um dos fetos no estágio 1 da doença, 80% no estágio 2, 65% no estágio 3 e 50% no estágio 4
Isoimunização de células vermelhas
Predição de anemia fetal
Em gestações com isoimunização de células vermelhas existe uma passagem transplacentária de anticorpos hemolíticos maternos que causam anemia fetal. O feto compensa a anemia com ajustes hemodinâmicos, mas quando o déficit de hemoglobina excede 6g/dL há o desenvolvimento de hidropsia fetal.
A anemia fetal é associada com diminuição da viscosidade sanguínea levando a aumento do retorno venoso e pré-carga com consequente aumento do débito cardíaco. Anemia fetal está associada com aumento da velocidade de fluxo arterial e venoso. A medida do pico de velocidade sistólica na artéria cerebral média (PVS-ACM) é um método útil para avaliar anemia fetal. Um ponto de corte no PVS-ACM de 1,5 DP da média, pode identificar quase todos os fetos severamente anêmicos com uma baixa taxa de falso positivo (cerca de 15%).
Medida do PVS-ACM também é útil na avaliação de fetos com outras causas de anemia como infecção por Prvovírus B19, hemorragia feto-materna e síndrome de transfusão feto-fetal.
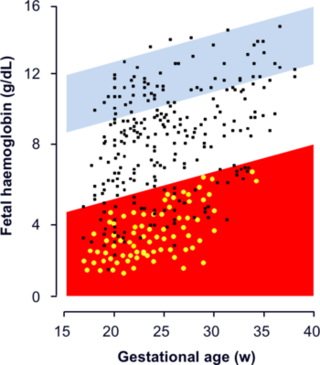
Manejo
O manejo em gestações com isoimunização tem com objetivo prever se o feto está gravemente afetado e corrigir a anemia fetal por transfusão sanguínea intrauterina. Em pacientes com gestações anteriores severamente afetadas e naquelas com níveis de anticorpos maternos maiores que 15 IU/mL (título 1:128 ou mais), o feto deve ser avaliado medindo-se o PVS-ACM em intervalos de 1-2 semanas iniciando a partir de 18 semanas. Cordocentese, para a medida da hemoglobina fetal, é realizada naquelas que desenvolvem ascites ou aumento do PVS-ACM.
Diabetes Mellitus
Comprometimento fetal
O comprometimento fetal em diabetes mellitus materno é determinado pelo tipo, duração e complicações maternas subjacentes
Em diabetes gestacional e em pré-gestacional não complicada (geralmente menos de 10 anos de duração) o fator de risco predominante é a hiperglicemia levando à macrossomia (geralmente a circunferência abdominal é proporcionalmente maior que a cabeça), polidramnia, cardiomegalia e morte fetal súbita.
Em diabetes pré-gestacional (geralmente mais de 10 anos de duração) com complicações renais e vasculares existe um risco aumentado de restrição de crescimento fetal (RCF) e pré-eclâmpsia. No entanto diferente da RCF por placentação inadequada, a RCF na diabetes pode ser acompanhada por aumento seguido de redução do líquido amniótico devido a hiperglicemia.
Acompanhamento Antenatal
O acompanhamento antenatal deve ser voltado para a detecção e monitoramento de:
Riscos mediados pela glicemia como macrossomia, polidrâmnio, e espessamento do miocárdio. O monitoramento é baseado em ultrassonografia semanal a partir do momento que as complicações tornam-se aparentes, o que acontece, geralmente, após 32 semanas. Se houver espessamento do miocárdio o parto deve ser considerado dependendo da idade gestcional
Riscos mediados por fatores vasculares como RCF e pré-eclâmpsia. O monitoramento é o mesmo que para outras causas de RCF e é baseado na combinação de (a) avaliação ultrassonográfica do crescimento fetal e líquido amniótico, (b) Doppler das artérias umbilicais, cerebral média e ducto venoso, e (c) monitoramento da frequencia cardíaca fetal. A frequência das avaliações depende da gravidade do comprometimento e varia de uma vez a cada duas semanas até várias vezes por semana.
Anticorpos Anti Ro/La
Bloqueio cardíaco fetal
Em bloqueio átrio-ventricular completo os átrios contraem no seu próprio rítmo, e nenhum dos seus impulsos são transmitidos para os ventrículos, que por sua vez tem uma frequência lenta (40-70 batimentos/min).
Em 50% dos casos estão presentes anomalias estruturais (mais frequentemente isomerismo esquerdo e transposição corrigida dos vasos da base). Nos demais casos, a condição é causada pela presença de auto-anticorpos maternos anti-Ro (SS-A) ou anti-La (SS-B) que atravessam a placenta e provocam lesão irreversível do miocárdio fetal e o sistema de condução. A maioria das gestantes são assintomáticas mas em alguns poucos casos, doença do tecido conjuntivo está presente (lúpus eritematoso, escleroderma, atrite reumatóide e síndrome de Sjogren). A prevalência de dano cardíaco fetal entre mulheres com anticorpos anti-Ro e anti-La é de 5-10%
O prognóstico depende da presença de defeitos cardíacos, frequência ventricular e presença de hidropsia, a qual aparece quando a frequência ventricular é menor que 50 batimentos/min
Em fetos com anomalias cardíacas o bloqueio cardíaco inicia no primeiro trimestre e o prognóstico é ruim
Bloqueio átrio-ventricular, secundário a auto-anticorpos maternos, desenvolve-se lentamente durante a gestação. Medidas de Doppler do intervalo PR permite a detecção de um bloqueio de primeiro grau (>130 ms). A administração materna de esteróides (dexametasona 8mg/dia) nesse estágio pode prevenir com sucesso a progressão para o bloqueio cardíaco completo. Uma vez que o bloqueio completo está estabelecido, nem dexametasona ou estimulação cardíaca tem sido bem sucedidas.
Taquicardia supraventricular
Taquicardia supraventricular
Taquicardia supraventricular é a forma mais comum de taquiarritimia. É caracterizada por frequência caríaca de 200-300 batimentos por minutos.
Taquicardia supraventricular pode ser causada por um foco autônomo, e nesse caso o rítmo é monotônico, ou pode ser causado por mecanismos de reentrada, e nesse caso é possível ver mudanças súbitas do ritmo de anormal para normal
Taquicardia persistente é associada com enchimento ventricular incompleto e diminuição do débito cardíaco. Isso resulta em sobrecarga atrial e insuficiência congestiva. Taquicardia persistente com frequência maior que 200 batimentos por minuto frequentemente resulta em hidropsia. A combinação de hidropsia e e disritmia tem prognóstico ruim com mortalidade de cerca de 80%
Avaliação do Doppler do ducto venoso durante a taquicardia supraventricular demonstra onda-a reversa com perda da característica trifásica da onda normal. O sinal mais precoce de sucesso do tratamento é o reaparecimento do fluxo trifásico, seguido do retorno ao normal do batimento cardíaco e finalmente a resolução da miocardiopatia pós taquicardia supraventricular
Gestação pós-termo
Gestação prolongada
Pós-termo. Em 5-10% das mulheres a gestação se continua além dos 294 dias (42 semanas completas) e são classificadas como pós-termo. Essas gestações tem risco aumentado para traumatismos no nascimento e asfixia, assim como chance aumentada de natimorto e morte pós-natal. É comumente aceito que a gravidez não deve ultrapassar 42 semanas e estudos randomizados demonstraram que a indução do parto às 41 semanas comparada com a conduta expectante , é associada com redução importante da mortalidade perinatal.
Pós-datismo refere-se a gestações além da data estimada do parto. Deve-se perceber que estas gestações também estão em risco aumentado de morte perinatal. De fato, o risco de perda da gestação (intrauterina, neonatal ou morte infantil) aumenta com a idade gestacional a partir de 38 semanas (1,2 por 1000 gestações em curso às 38 semanas para 2,4 às 40 semanas e 5,8 às 43 semanas).
- O risco de natimorto está relacionado ao envelhecimento placentário, porém não existe uma sequência específica de piora da condição fetal para guiar o acompanhamento fetal. No entanto, existe uma rápida diminuição do volume amniótico e a vigilância deve ser baseada na avaliação do líquido amniótico duas vezes por semana.
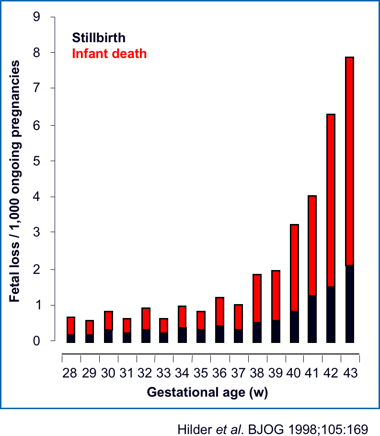
Acompanhamento em condições específicas
Conclusão
- A forma de acompanhamento deve ser específico para cada doença
- A frequência dos testes deve ser baseada na evolução da doença
- Os limites de intervenção devem ser baseados nos riscos intrauterinos versus neonatais
Agradecimentos
Lecturer: Ahmet Baschat
Director: Kypros Nicolaides
Multimedia: Nicola Persico
Filming: Jader de Jesus Cruz
Programming: Chris Harris
Translators:
- Polish: Rafa? Kocy?owski
- Portuguese: Jader de Jesus Cruz, James Murray Anderson e Carolina Ribeiro
- Russian: Elena Emelyanenko
- Spanish: Jose Moratalla
- Vietnamese: Nguyen Ha
Educação
A varredura de 11-13 semanas
Curso pela internet
Este curso cobre todos os aspectos importantes da triagem no primeiro trimestre para anomalias cromossômicas e defeitos importantes.
- O curso é gratuito
- Está disponível em vários idiomas
- O curso completo dura várias horas
- Não precisa ser abordado em uma única sessão
- Se você quiser ver o curso todo ou partes de seu interesse, clique aqui .
- O curso é obrigatório para todos os ultrassonografistas que desejam obter os Certificados da FMF de competência na medição da translucência nucal, avaliação do osso nasal, avaliação do fluxo do ducto venoso e avaliação do fluxo tricúspide. Se você deseja participar do curso, faça o login aqui .
Reserve na varredura de 11-13 semanas
Este livro, que é gratuito, resume todos os aspectos do rastreamento de anormalidades cromossômicas e estruturais por meio da varredura de 11-13 semanas
Três créditos de Educação Médica Continuada
Depois de concluir o curso, você pode obter um certificado CME . Para os termos e condições, clique aqui.
Lista de inscritos
Para ver a lista de profissionais de saúde que concluíram o curso baseado na Internet na varredura de 11-13 semanas, clique aqui.
Ultrassonografia de 11-13 semanas
Bem vindo a este curso educacional a sobre ultrassonografia (US) de 11-13 semanas da Fetal Medicine Foundation
A Fetal Medicine Foundation, uma organização sem fins lucrativos, trabalha para o benefício das mulheres grávidas e suas famílias. Nós:
- Financiamos e promovemos pesquisas para melhorar o diagnóstico pré-natal
- Promovemos formação e treinamento para médicos de todo o mundo
- Fornecemos informações atualizadas para pacientes e profissionais de saúde
O método tradicional de rastreio da síndrome de Down era baseado na idade materna, sendo a amniocentese ou a biópsia de vilo corial oferecidas às grávidas com idade igual ou superior à 35 anos. Isto resultava na necessidade de testes invasivos em 15-20% das grávidas com uma taxa de detecção inferior a 50% dos fetos com síndrome de Down, uma vez que a maioria dos fetos afetados vem de um grupo de grávidas mais jovem
Um método mais eficiente de rastreio é baseado na combinação de:
- Idade materna
- Quantificação de produtos placentários – ß-hCG livre e PAPP-A – no sangue materno
- Ultrassonografia entre as semanas 11-13 da gestação para:
- medir a quantidade de líquido na parte posterior da nuca (translucência nucal)
- avaliar o nariz e palato fetal
- medir a freqüência cardíaca fetal
- avaliar o fluxo sanguíneo através da válvula tricúspide e do ducto venoso
Este novo método de rastreio reduz dramaticamente o número de grávidas que necessitam de um teste invasivo, de aproximadamente 20% para menos de 3% e ao mesmo tempo aumenta a taxa de detecção de síndrome de Down e outras cromossomopatias de menos de 50% para mais de 95%
Outros benefícios do US entre 11–13 semanas são:
- Datar a gestação com maior acuidade
- Diagnosticar precocemente várias cromossomopatias
- Detecção de gestação múltipla e diagnóstico confiável da corionicidade, que é o principal determinante do prognóstico destas gestações
- Outra vantagem recentemente descrita é a de identificar as grávidas com maior risco para desenvolver pré-eclâmpsia
Apesar de existirem múltiplos benefícios no US das 11-13 semanas, é essencial que aqueles que realizam o exame tenham um bom conhecimento dos critérios diagnósticos e manejo clínico das condições identificadas nesse exame, estejam adequadamente treinados para realizar esse tipo de exame com um alto padrão de qualidade, e submetam os seus resultados a um controle de qualidade baseado na distribuição das medidas e avaliação das imagens obtidas
Esperamos que você considere as informações aqui fornecidas proveitosas sendo sugestões para melhorar este curso bem vindas
Ultrassonografia de 11-13 semanas
Amniocentese e biópsia de vilo corial
Rastreio de cromossomopatias
Diagnóstico das anomalias fetais maiores
Rastreio e diagnóstico de gemelaridade
Rastreio de pré-eclâmpsia
Diagnóstico de cromossomopatias
Amniocentese e Biópsia de Vilo Coral (BVC)
- O diagnóstico das cromossomopatias requer testes invasivos como a amniocentese ou a biópsia de vilo corial (BVC)
- Estudos randomizados têm demonstrado que o risco de aborto devido à BVC no primeiro trimestre é o mesmo da amniocentese no segundo trimestre, que é cerca de 1%
- A amniocentese não deve ser realizada antes de 16 semanas pois com a amniocentese precoce a taxa de aborto é cerca de 2% maior e a incidência de talipes equinovarus é 1,5% maior que a BVC no primeiro trimestre ou a amniocentese no segundo trimestre
- A BVC não deve ser realizada antes das 11 semanas pois a BVC precoce está associada a defeitos transversos dos membros fetais, micrognatia e microglossia
- Os procedimentos invasivos devem ser realizados por profissionais apropriadamente treinados e experientes. Evidências recentes sugerem que com tais profissionais, o risco de aborto pode ser tão baixo quanto 1 em 1000.
Idade materna
Translucência nucal
Frequência cardíaca fetal
Avaliação bioquímica
Novos marcadores ultra-sonográficos
Rastreio das cromossomopatias
Princípios gerais
- Todas as mulheres tem o risco de ter um feto/bebê com defeitos cromossômicos
- O risco basal ou risco a priori depende da idade materna e gestacional
- O risco individual (patiente-específico) é calculado multiplicando o risco basal por uma série de riscos relativos , que dependem dos resultados de uma série de testes de rastreio
- O risco relativo para uma dada medida ecográfica ou bioquímica é calculada através da divisão da percentagem de fetos cromossomicamente anormais pela percentagem de fetos normais com a mesma medida
- Sempre que um teste é realizado, o risco basal é multiplicado pelo risco relativo desse teste para se calcular um novo risco, que então se torna o risco basal para o próximo teste
- Se os testes não forem independentes, então técnicas mais sofisticadas, envolvendo estudos estatísticos de análise multivariável, podem ser usadas para se calcular o risco relativo combinado
Idade materna
Risco de trissomia 21
- Aumenta com idade materna
- Diminui com a idade gestacional pois cerca de 30% dos fetos afetados morrerão entre 12 e 40 semanas de gestação
Por exemplo, em mulher com 35 anos de idade, o risco para trissomia 21 com 12 semanas de gestação é de 1 em 250, mas a chance de ela parir um bebê afetado com 40 semanas, é de 1 em 350.
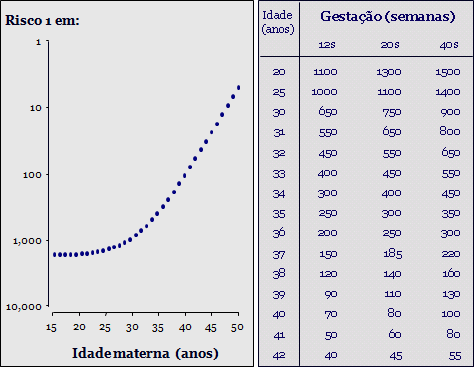
Idade materna
Outras aneuploidias
- O risco para trissomias 18 e 13 aumenta com a idade materna e diminui com a idade gestacional. A taxa de óbito fetal entre 12 e 40 semanas é cerca de 80%
- A sindrome de Turner não está relacionado com a idade materna. A taxa de óbito fetal entre 12 e 40 semanas é de cerca de 80%. A prevalência é de aproximadamente 1 em 1500 às 12 semanas e 1 em 4000 às 40 semanas
- A triploidia não está relacionada com a idade materna. A prevalência às 12 semanas é aproximadamente 1 em 2000, mas é altamente letal e raramente observada entre os nativivos
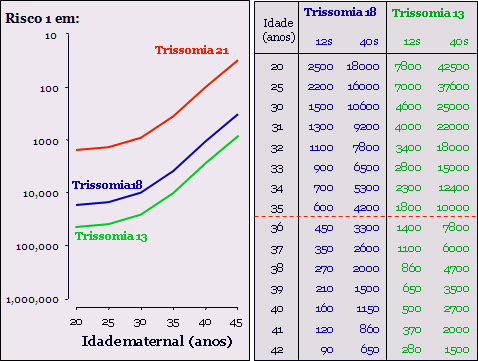
Idade materna
Performance do rastreio para trissomia 21
O risco de trissomia 21 aumenta com a idade materna. Contudo como existem mais mulheres no grupo mais jovem, a maioria dos fetos com trissomia 21 vêm do grupo de mulheres abaixo dos 35 anos de idade
Nas décadas de 70 e 80 o rastreio para trissomia 21 era basedo na idade materna e a amniocentese ou BVC eram oferecidos se a idade era igual ou superior a 35 anos. Como cerca de 5% das grávidas tinham 35 anos ou mais, a política de rastreio baseado na idade materna resultava em:
-
- Taxa de testes invasivos 5%
- Taxa de detecção de trissomia 21 30%
Na maioria dos países desenvolvidos nos últimos 30 anos a idade materna tem aumentado e agora cerca de 20% das gestações, incluindo 50% dos fetos com trissomia 21, ocorrem em mulheres com 35 anos ou mais.
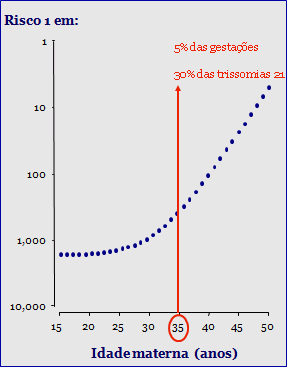
Translucência Nucal
Definição
- Translucência nucal (TN) é a representação sonográfica da coleção de líquido sob a pele, atrás da nuca, no primeiro trimestre da gestação.
- A expressão “ translucência” é usada quando existe ou não septação e quando é restrita à nuca ou envolve o feto inteiro.
- A incidência de cromossomopatias e outras anormalias está relacionada com o tamanho e não com a aparência da TN.
- Durante o segundo trimestre a TN geralmente resolve espontaneamente e, em alguns casos pode evoluir para edema da nuca ou higroma cístico, acompanhado ou não de hidropsia fetal.
Idade Gestacional
A idade gestacional ideal para a medida da TN fetal e de 11+0 a 13+6 semanas. O comprimento cabeça-nádega (CCN) mínimo deve ser 45 mm e o máximo 84 mm
As razões para escolher 11 semanas como a idade gestacional mínima são:
- Os testes de rastreio necessitam de correspondentes testes diagnósticos disponíveis e a BVC antes de 11 semanas está associada a defeitos transversos dos membros fetais
- Muitas das anormalias fetais maiores podem ser diagnosticadas durante a ultrassonografia de TN, quando da idade gestacional mínima de 11 semanas
As razões para escolher 13 semanas e 6 dias como a idade gestacional maxima são:
- Possibilitar às mulheres, com fetos acometidos, a opção de interrupção da gestação no primeiro trimestre ao invés de no segundo trimestre.
- A incidência de um acúmulo anormal de líquido atrás da nuca em fetos com defeitos cromossômicos, diminui depois de 13 semanas
- A taxa de sucesso em medir corretamente a TN, diminui após 13 semanas uma vez que o feto tende a permanecer em posição mais verticalizada tornando mais difícil a obtenção da imagem apropriada
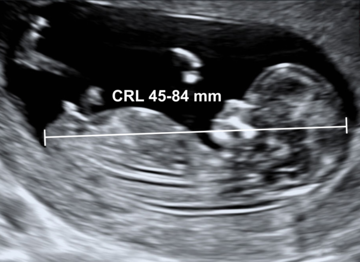
Medida
A magnificação da imagem deve ser suficiente para a que apenas a cabeça e a parte superior do tórax ocupem toda a tela
- Deve ser obtido um corte sagital do feto. Nesse corte, o osso nasal ecogênico e o palato retangular são vistos separadamente. Rotação da cabeça resulta em não visualização da ponta do nariz e aparecimento do osso maxilar com estrutura ecogênica entre osso nasal acima e a parte anterior do palato abaixo. Com rotação maior, o osso nasal desaparece e há aumento do osso maxilar coalescendo com o palato.
- O feto deve estar em uma posição neutra e a cabeça alinhada com a coluna vertebral. Se o pescoço fetal estiver extendido a medida estará falsamente aumentada e se estiver fletida a medida estará falsamente diminuída
- É preciso diferenciar atentamente a linha da pele fetal com o âmnio
Medida
O ponto mais largo da TN é o que sempre deve ser medido
- A medida deve ser feita utilizando o bordo interno da linha horizontal do caliper colocada SOBRE o bordo da linha que define a transluscência nucal – a barra horizontal do caliper deve ficar de uma forma que seja difícil a visualização já que se funde à linha branca da TN e não ao líquido
- Ao se magnificar a imagem ( pré ou pós freeze zoom) é importante diminuir o “ganho” do aparelho. Isso evita o erro de medir a TN com uma linha sem definição o que torna a medida imprecisa e muitas vezes subestimada
- Durante o exame, a medida da TN deve ser verificada mais de uma vez, e a maior medida, que respeita os critérios acima descritos, é a que deve ser utilizada.
Medida
Circular cervical de cordão pode ocorrer em 5% dos casos e a medida da TN pode parecer falsamente aumentada.
Nesses casos, a medida da TN acima e abaixo do cordão são diferentes, então, o cálculo do risco deve usar a média entre as duas medidas.
Rastreio para trissomia 21
Em fetos euplóides, a espessura da TN aumenta com oCCN.
Em 75-80% das trissomias 21 a TN esta acima do percentil 95 na curva de normalidade
Nos fetos com trissomia 21 não existe relação com espessura da TN e idade materna
A idade materna pode ser combinada com a medida da TN para um rastreio mais eficaz das cromossomopatias no primeiro trimestre
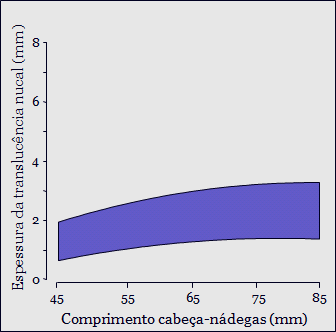
Risco relativo
Em um feto com determinado CCN, cada medida da TN representa um risco relativo, o qual é multiplicado pelo risco basal materno e ligado à idade gestacional para calcular um novo risco.
Quanto maior a TN, maior será o risco relativo e por sua vez, maior o novo risco.
Quanto menor a TN, menor será o risco relativo e por sua vez, menor será o novo risco
O risco é maior em uma mulher de 20 anos de idade com uma medida de TN aumentada do que em uma mulher de 40 anos com uma TN pequena.
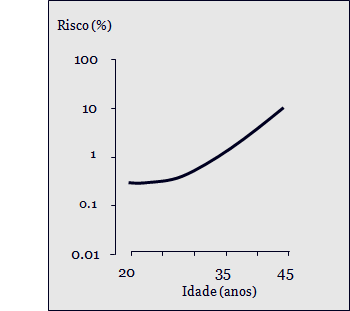
Modelo misto
Na distribuição da espessura da TN em fetos euplóides:
- A mediana e os percentis 1,5 e 95 aumentam com o CCN
- O percentil 99 é em torno de 3,5mm e não varia com o CCN
A melhor forma de explicar este achado é considerar que a TN fetal segue uma mescla de duas curvas:
- Uma é CCN dependente
- Outra que é CCN independente

Modelo misto
A distribição onde a TN aumenta de acordo com o CCN é a mesma para os fetos cromossomicamente anormais e euplóides, todavia, a proporção que segue essa distribuição é grande no grupo euplóide ( cerca de 95%) e pequena no grupo anormal, sendo cerca de 5%, 30%, 15% e 20% para as trissomias 21, 18, 13 e Turner respectivamente
A distribuição em que a TN não muda de acordo com o CCN é em primeiro lugar, pequena para os fetos euploides e grande para o grupo anormal. E em segundo lugar a TN média é diferente, sendo 2,0 mm para o grupo euploide e 3,4 mm, 5,5 mm, 4,0 mm e 9,2 mm para as trissomias 21, 18, 13 e Turner respectivamente
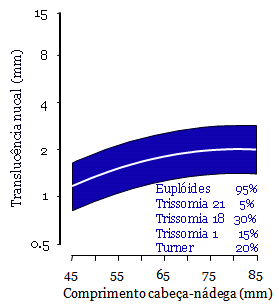
Frequência cardíaca fetal
Medida
- É necessário um corte transverso ou longitudinal do coração fetal
- Doppler pulsátil é usado para obter 6-10 ciclos cardíacos durante um período de repouso fetal
- A frequência cardíaca fetal é calculada pelo software do próprio aparelho de ecografia
Fetos euplóides e trissômicos
Em fetos euplóides, a FCF aumenta de cerca de 110 bpm na 5ª semana para 170 bpm na 10ª semana e então, diminui gradualmente até 150 na 14ª semana.
Na trissomia 21, a FCF é levemente aumentada e acima do percentil 95 em cerca de 15% dos casos.
Na trissomia 18, a FCF é levemente diminuída e abaixo do percentil 5 em cerca de 15% dos casos.
Na trissomia 13, a FCF é substancialmente aumentada e acima do percentil 95 em 85% dos casos.
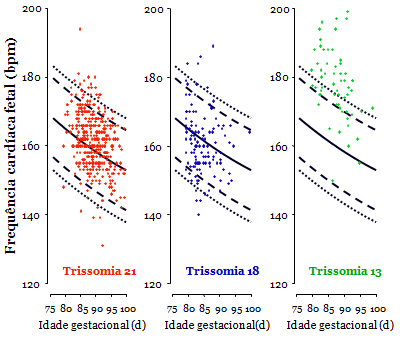
Rasteio para trissomias
A inclusão da FCF no rastreio para cromossomopatias de primeiro trimestre, combinado com a ecografia e bioquímica , tem um impacto pequeno para a detecção das trissomias 21 e 18, contudo, tem um grande impacto na detecção da trissomia 13
Além disso, a inclusão da FCF é importante para diferenciar entre trissomia 18 e 13, as quais são similares na sua apresentação com TN aumentada e baixa dosagem de ?-hCG livre e PAPP-A no sangue materno

Avaliação bioquímica
Rastreio para trissomia 21
Gestações com trissomias são associadas a concentrações alteradas de vários produtos feto-placentários no sangue materno
Rastreio no segundo trimestre baseado na idade materna e combinações de ß-hCG livre ou total, AFP, uE3 and Inibina A podem identificar 56-71% das trissomias 21 com uma taxa de falso positivo de 5%.
Rastreio no 1º trimentre com a combinação de idade materna, TN fetal, FCF com ß-hCG livre e PAPP-A é capaz de identificar cerca de 90% das trissomias 21 com uma taxa de falso positivo de 3%.
Ajuste de medidas
A medida da concentração sérica de ß-hCG livre e PAPP-A é influenciada pelo equipamento e reagentes usados, pela idade gestacional e peso materno, etnicidade, tabagismo e método de concepção
No cálculo de risco paciente-específico, são necessários ajustes para a medida de ß-hCG livre e PAPP-A. Cada medida é primeiro convertida em um múltiplo da mediana normal estimada (MoM), específico à uma gestação com as mesmas características de idade gestacional, peso materno, tabagismo, etnicidade e método de concepção
- Em mulheres afro-descendentes, os níveis de PAPP-A são cerca de 60% mais elevados do que nas mulheres brancas. Não levar em consideração a origem étnica resultaria em uma substancial subestimação do real risco de trissomia 21 em mulheres afro-descendentes
- Nas mulheres que fumam e aquelas e engravidam por métodos como FIV, o nível sérico de PAPP-A é mais baixo e isso pode superestimar o risco de trissomia 21, e gerar um substancial aumento da taxa de falso positivo
Rastreio para trissomia 21
Nas trissomias 21, o ß-hCG livre no soro materno está aproximadamente duas vezes mais elevado e PAPP-A está reduzido pela metade, comparado com níveis em gestações cromossomicamente normais
A performance do rastreio para trissomia 21 baseado na idade materna e na concentração sérica de ß-hCG livre e PAPP-A é:
- Taxa de Detecção 65%
- Taxa de Falso Positivo 5%
Rastreio para trissomia 21
Nas gestações com trissomia 21, o ß-hCG livre é mais elevado que nas euplóides e a diferença entre as duas é maior na 13ª do que na 11ª semana
Nas gestações com trissomia 21, o nível sérico de PAPP-A é mais baixo do que nas euplóides, e a diferença entre as duas é maior na 11ª do que na 13ª semana
A diferença dos euplóides dos níveis no PAPP-A na 11ª semana é maior que a diferença no ß-hCG na 13ª semana. Sendo assim a performance do rastreio bioquímico é melhor na 11ª do que na 13ª semana
Rastreio para outras aneuploidias
Nas gestações euplóides a média de ß-hCG livre é 1.0 MoM e PAPP-A é 1.0 MoM
Nas gestações aneuplóides a média de valores de MoM de ß-hCG livre e PAPP-A é:
| ß-hCG livre | PAPP-A | |
|---|---|---|
| T21 | ||
| T18 | ||
| T13 | ||
| Turner | ||
| Triploidia | ||
| Materna | ||
| Paterna |
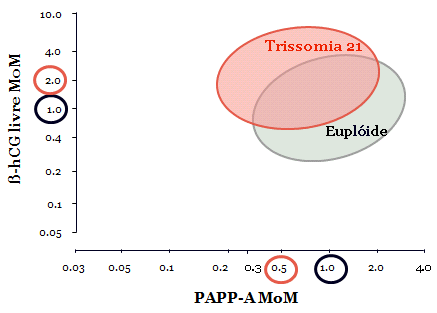
Rastreio combinado
Trissomia 21
Gestações com trissomias 21 comparadas às euplóides:
- A diferença nos marcadores bioquímicos é maior na 11ª do que na 13ª semana
- A diferença na TN é maior na 11ª do que na 13ª semana
- Consequentemente, a performance do screening é melhor na 11ª do que na 13ª semana
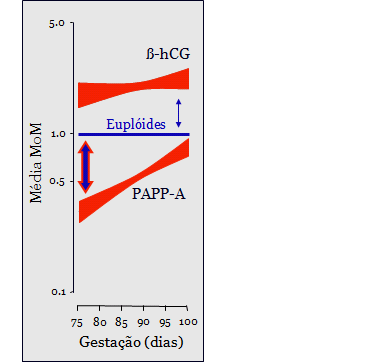
Efeito da idade gestacional
A performance geral do rastreio combinado é melhor na 11ª do que na 13ª semana e pode ser melhor ainda na 10ª semana
A ultrassonografia para identificar anomalias fetais é melhor na 12ª do que na 11ª semana e é ainda muito melhor no que na 10ª semana
Uma boa forma de alcançar uma alta performance no rastreio de trissomia 21 e diagnosticar, em conjunto, defeitos fetais é realizar o teste bioquímico na 10ª ou 11ª semana e a ultrassonografia na 12ª semana
Trissomia 18 e 13
As trissomias 18 e 13 são a segunda e terceira cromossomopatias mais comuns depois da trissomia 21.
Entre 11 -13 semanas a prevalência das trissomias 18 e 13 comparando com trissomia 21 é de cerca de 1:2.5 and 1:7, respectivamente.
Todas as 3 trissomias são associadas a idade materna avançada, TN fetal aumentada, e níveis séricos de PAPP-A diminuidos
Uma consequência benéfica do método combinado de rastreio para trissomia 21 é o diagnóstico precoce das trissomias 18 e 13. Para uma taxa de falso positivo de 3% a taxa de detecção de trissomia 21 é de 90% e para trissomias 18 e 13 é cerca de 75%
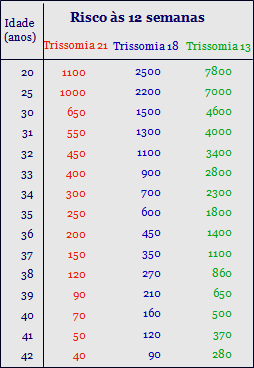
Trissomia 18 e 13
Diferenças entre as trissomias 21, 18 e 13:
- TN fetal é maior em trissomias 18 e 13 do que em trissomia 21.
- Níveis séricos de PAPP-A são menores nas trissomias 18 e 13 do que na trissomia 21.
- Níveis séricos de ß-hCG livre na trissomia 21 são altos enquanto na trissomia 18 e 13 são baixos.
- Frequência cardíaca fetal na trissomia 13, ao contrário das trissomias 21 e 18, é elevada.
O uso de algorítmos específicos para trissomias 18 e 13, acrescentados do algorítmo para trissomia 21, melhora a detecção das trissomias 18 e 13 de 75% para 95% com um aumento mínimo da taxa de falso positivo 3% para 3.1%.
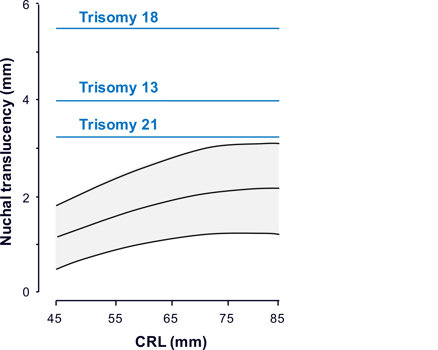
Osso Nasal
Fluxo no Ducto Venoso
- Cromossomopatias
- Defeitos cardíacos maiores
- Óbito Fetal
Fluxo pela Válvula Tricúspide
- Cromossomopatias
- Defeitos Cardíacos maiores
Novos marcadores ultrassonográficos
Visão geral
Um rastreio eficaz de trissomias no 1º trimestre é composto pela combinação de idade materna, TN fetal, frequência cardíaca fetal e níveis séricos de ß–hCG e PAPP-A no soro materno.
- A avaliação dos novos marcadores melhora o desempenho do teste combinado, aumentando a taxa de detecção e diminuindo a taxa de falso positivo.
- A avaliação dos novos marcadores requer treino apropriado dos profissionais e certificação da sua capacidade em realizar esse tipo de exame.
Novos marcadores ultrassonográficos
Estratégias de rastreio
Existem 2 estratégias para o uso dos novos marcadores no rastreio de trissomia 21, com taxas de detecção e de falso positivo semelhantes:
- Avaliar todos ou alguns dos novos marcadores, em todos os casos.
- Novos marcadores são avaliados somente no sub-grupo de gestações com um risco intermediário ( entre 1 em 51 até 1 em 1000) após o teste de rastreio combinado (TN, FCF, ß-hCG livre e PAPP-A), constituindo somente um sexto da população total.
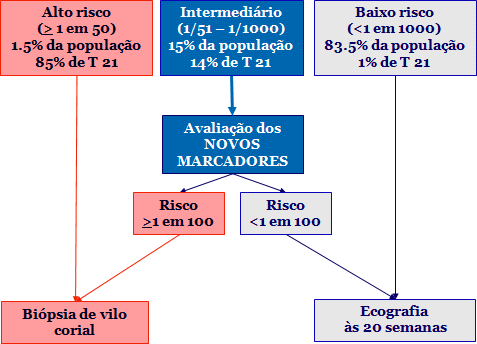
Osso nasal
Critérios
Para avaliar o osso nasal a gravidez deve estar entre 11+0-13+6 semanas e o CCN entre 45-84 mm.
- A magnificação da imagem deve ser a sufuciente para que somente a cabeça e a porção superior do tórax ocupem toda a tela.
- Um corte sagital médio do perfil fetal deve ser obtido.
- O transdutor deve estar paralelo ao osso nasal e a sonda deve ser gentilmente inclinada a fim de varrer de um lado ao outro o nariz fetal.
- Quando os critérios forem preenchidos, três linhas distintas devem ser visualizadas a nível do nariz fetal:
- A linha superior representa a pele.
- A inferior, que é mais grossa e mais ecogênica que a pele acima, representa o osso nasal.
- Uma terceira linha na frente do osso porém mais apical que a pele representa a ponta do nariz.
- O osso nasal é considerado presente quando for mais ecogênico do que a pele acima dele; e é considerado ausente quando a ecogenicidade for a mesma ou menor que a da pele ou quando o osso não for visualizado.
Rotação da cabeça em cerca de 100 da linha média resulta na não visualização da ponta do nariz e aparecimento do osso maxilar com estrutura ecogênica entre osso nasal acima e parte anterior do palato abaixo. Com maior rotação, o osso nasal desaparece e há aumento do osso maxilar e coalescência com o palato.
Osso nasal ausente
Entre 11-13 semanas o osso nasal é considerado ausente em cerca de:
-
- Fetos euplóides 1-3%
- Fetos com trissomia 21 60%
- Fetos com trissomia 18 50%
- Fetos com trissomia 13 40%
Osso nasal ausente é mais comum se:
- A gestação estiver na 11ª semana do que na 13ª .
- A transluscência nucal está aumentada.
- Afro-descendentes.

Estratégias de rastreio
Existem 2 estratégias de avaliação do osso nasal no rastreio da trissomia 21 com taxas de detecção e falso positivo semelhantes:
- O osso nasal é examinado em todos os casos.
- O osso nasal é examinado somente no subgrupo de gestações com um risco intermediário depois do teste combinado (TN, FCF, ß-hCG livre e PAPP-A), que constitui apenas um sexto da população total.
A avaliação do osso nasal melhora o desempenho do rastreio combinado, aumentando a taxa de detecção de 90% para 93% e diminuindo a taxa de falso positivo de 3% para 2.5%
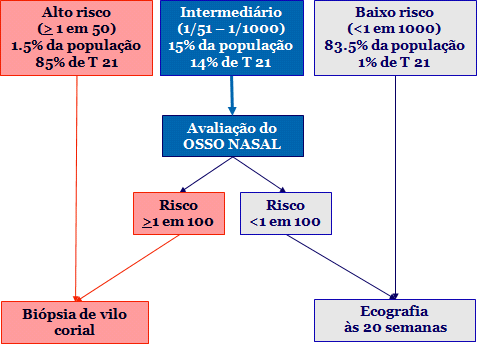
Fluxo do ducto venoso
Visão geral
O ducto venoso é um vaso curto que conecta a veia umbilical com a veia cava inferior
- O ducto venoso tem um papel importante na circulação fetal desviando o sangue oxigenado preferencialmente para o cérebro.
- Cerca de 80% do sangue oxigenado vindo da placenta, é desviado do fígado e é direcionado ao coração. O sangue entra no átrio direito e passa para o átrio esquerdo pelo forâmen oval. Do átrio esquerdo, o sangue passa para o ventrículo esquerdo e daí para aorta.
- O ducto venoso geralmente fecha alguns minutos após o nascimento, contudo em neonatos prematuros isso pode demorar um pouco mais.
Critérios
Para avaliação do ducto venoso a idade gestacional deve ser entre 11+0-13+6 semanas e o CCN entre 45-84 mm
- O feto não pode estar em movimento.
- A magnificação da imagem deve ser o suficiente para que, somente o tórax e o abdomen fetal ocupem toda a tela.
- Um corte sagital médio à direita do tronco fetal deve ser obtido.
- Deve ser usado o Doppler colorido para identificar a veia umbilical, ducto venoso e coração fetal.
- O volume de amostra no doppler pulsátil deve ser pequeno (0.5-1.0 mm) a fim de evitar contaminação com vasos adjacentes e o mesmo deve ser colocado na área de aliasing (amarelada).
- O ângulo de insonação deve ser menor que 30 graus.
- O filtro deve ser calibrado com uma frequência baixa (50-70 Hz) a fim de permitir a visualização de toda a onda.
- A velocidade da onda (sweep speed) deve ser alta (2-3 cm/s) a fim de a onda de fluxo estar amplamente estendida para uma melhor avaliação da onda “a”.
Fluxo normal e anormal
O fluxo sanguíneo no ducto venoso tem um formato de onda característico com:
- Alta velocidade durante a sístole ventricular (onda S) e diástole (onda D).
- Fluxo anterógrado durante a contração atrial (onda A).
Avaliação qualitativa do fluxo sanguíneo no ducto venoso é baseada na aparência da onda A.
- Positiva ou ausente (normal).
- Reversa (anormal).
Contaminação do formato de onda
Avaliação qualitativa do fluxo sanguíneo pelo ducto venoso é baseado na aparência da onda A:
- Positivo ou ausente (normal)
- Reverso (anormal)
A onda é contaminada com fluxo de vasos adjacentes quando volume de amostra do doppler pulsátil:
- Estiver maior que o necessário (>1.0 mm)
- Não estiver colocado precisamente no ducto venoso

Onda A reversa
Entre 11-13 semanas onda A reversa pode ser encontrada em aproximadamente:
- Fetos euploides 3%
- Fetos com trissomia 21 65%
- Fetos com trissomia 18 55%
- Fetos com trissomia 13 55%
Onda A reversa é mais comum se:
- A gestação estiver na 11ª semana do que na 13ª.
- A transluscência nucal está aumentada.
- Nível de PAPP-A estiver reduzido.
- Afro-descendentes.
Onda A reversa é associada com aumento do risco para:
- Anomalias cromossômicas
- Defeitos cardíacos
- Morte fetal
Entretanto em cerca de 80% dos casos com onda A reversa o desfecho gestacional é normal.
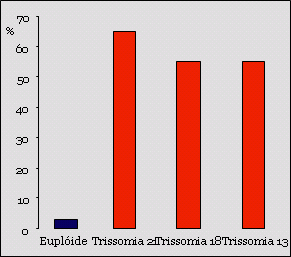
Estratégias de rastreio
Existem 2 estratégias para avaliação do ducto venoso no rastreio de trissomia 21, ambas com taxas de detecção e falso positivo semelhantes:
- O ductos venoso é utilizado em todos os casos, com a vantagem não somente de melhorar o desempenho do screening de cromossomopatias como também identificar gestações com risco aumentado de defeitos cardíacos e morte fetal.
- O Ducto venoso é utilizado somente no sub-grupo de gestantes com um risco intermediário depois do teste combinado (TN, FCF, ß-hCG livre e PAPP-A) que constitui apenas um sexto da população total.
Avaliação do ducto venoso melhora a performance do teste combinado aumentando a taxa de detecção de 90% para 95% e diminuindo a taxa de falso positivos de 3% para 2,5%.

Defeitos cardíacos
A prevalência de defeitos cardíacos maiores em fetos euplóides é aproximadamente 4 em 1,000
- O risco de defeitos cardíacos maiores está aumentado quando a TN está aumentada.
Nos fetos com TN aumentada, o risco de defeitos cardíacos maiores é modificado por achados do ducto venosos:
-
- Aumentado se a onda A estiver reversa.
- Diminuído de a onda A estiver normal.
Se a onda A do ducto venoso é reversa, é importante realizar ultrassonografia detalhada para excluir ou diagnosticar defeitos cardíacos maiores.
Morte fetal
O risco de abortamento entre 11 semanas e o nascimento é cerca de 2%
A prevalência de onda A reversa entre 11-13 semanas é mais de 10% nas gestações que resultam em perda fetal e menos que 4% nas que resultam em nascidos-vivos.
O risco de morte fetal está aumentado quando:
-
- O ducto venoso tem onda A reversa.
- O nível sérico de PAPP-A está baixo.
- A gestante é afro-descendente.
- A mãe é obesa.
Para Ducto venoso reverso:
-
- Monitorizar o crescimento fetal (US nas 20ª, 28ª e 34